

ANTI-ADIPOGENIC (Adipocyte Differentiation Inhibitor)
March 16, 2020
GYNOSTEMMA PENTAPHYLLUM (The Herb of Immortality)
April 27, 2020
SENOLYTIC (Zombie Cell Killer)
$275.00
SENOLYTIC
ZOMBIE CELL KILLER!
200:1 Concentration
Understanding Zombie Cells: The Role of Senescence and Senolytic Therapies in Health and Longevity
As we age, our bodies undergo a myriad of changes, one of which involves the accumulation of senescent cells, often referred to as “zombie cells.” These cells have sustained damage and enter a state of senescence, meaning they stop dividing but resist programmed cell death. This phenomenon is linked to various age-related diseases, including Alzheimer’s, osteoarthritis, and cardiovascular dysfunction.
What Are Senescent Cells?
Senescent cells are characterized by irreversible growth arrest and a unique secretory profile known as the senescence-associated secretory phenotype (SASP). This profile includes the release of pro-inflammatory cytokines, growth factors, and proteases that can disrupt tissue function and induce inflammation, leading to neighboring healthy cells’ dysfunction. Accumulation of these senescent cells over time contributes to what is termed “inflammaging,” a chronic low-grade inflammation associated with aging and age-related diseases.
The Senescence-Associated Secretory Phenotype (SASP)
The SASP plays a crucial role in the detrimental effects of senescent cells. While initially a mechanism for tumor suppression, the SASP can promote chronic inflammation and tissue degeneration. This dual role creates a complex relationship where senescent cells can support wound healing but also contribute to the onset of diseases like cancer, atherosclerosis, and neurodegenerative conditions. Understanding the SASP is vital for developing therapeutic strategies targeting senescent cells.
Senolytic Therapies: Targeting Zombie Cells
Senolytics are a class of drugs designed to selectively induce apoptosis, or programmed cell death, in senescent cells. Research has shown that eliminating these cells can improve health outcomes and potentially extend healthspan—the period during which an individual is generally healthy and free from serious disease. Various compounds, such as dasatinib and quercetin, have been identified as effective senolytics, showing promise in preclinical studies with mice. These therapies can alleviate symptoms associated with frailty, vascular aging, chronic hypercholesterolemia, and metabolic disorders like type 2 diabetes.
Mechanisms of Senolytics: Key Pathways Involved
The efficacy of senolytic therapies is linked to several biological pathways and factors that regulate cell survival and apoptosis:
1. BCL-2 Family Proteins: Proteins such as BCL-2 and BAX play pivotal roles in regulating apoptosis. Senolytics can target these pathways to promote the death of senescent cells.
2. PI3K/AKT Pathway: This pathway is crucial for cell survival and growth. Inhibition of this pathway has been shown to enhance the sensitivity of senescent cells to apoptosis.
3. Nrf2 and Autophagy: Nrf2 is a transcription factor that protects cells from oxidative stress, while autophagy helps remove damaged cellular components. Targeting these pathways may enhance the clearance of senescent cells.
4. mTOR and Wnt Signaling: The mTOR pathway is associated with cell growth and metabolism, while Wnt signaling plays a role in cell fate decisions. Both pathways can influence the viability of senescent cells.
5.cGAS-STING Pathway: This pathway responds to cytosolic DNA and is involved in immune responses. Senescent cells can activate this pathway, leading to inflammation and potential tissue damage.
6. Caspases and Other Pro-Apoptotic Factors: Caspases are essential for the execution of apoptosis. Senolytic therapies may promote the activation of these molecules to effectively eliminate senescent cells.
Clinical Implications and Future Directions
The potential of senolytic therapies extends beyond merely prolonging lifespan; they aim to enhance healthspan by treating age-related conditions. Clinical trials are underway to assess the safety and efficacy of these drugs in humans. The goal is to identify effective biomarkers for senescent cells, allowing for personalized approaches to treatment.
There is a growing interest in understanding the balance between the beneficial and harmful effects of senescent cells. While they play a role in tissue repair and tumor suppression, their accumulation can lead to detrimental consequences. Therefore, a nuanced approach to targeting senescent cells is necessary to maximize therapeutic benefits while minimizing potential risks.
Conclusion: The Future of Aging Research
As research continues to unravel the complexities of senescent cells and their role in aging, senolytic therapies represent a promising frontier in the quest for healthy longevity. By targeting the mechanisms underlying cellular senescence, we may be able to mitigate the adverse effects associated with aging, paving the way for a healthier, more active life in later years.
Understanding the relationship between senescent cells, the SASP, and senolytic pathways can provide insight into the aging process and the development of targeted therapies. As science progresses, the hope is to transform our approach to aging, not merely as an inevitable decline but as a process that can be managed and improved through innovative medical interventions.
THE KEY TO ELIMINATING ZOMBIE SENESCENT CELLS CONSISTS OF TWO PARTS:
1. DOWN-REGULATING ANT-APOPTOTIC PATHWAYS THAT PROTECT THEM
2. UP-REGULATING PRO-APOPTOTIC PATHWAYS THAT CAN KILL THEM
WHICH PATHWAYS?
HERE’S AN EXAMPLE OF SOME OF THEM:
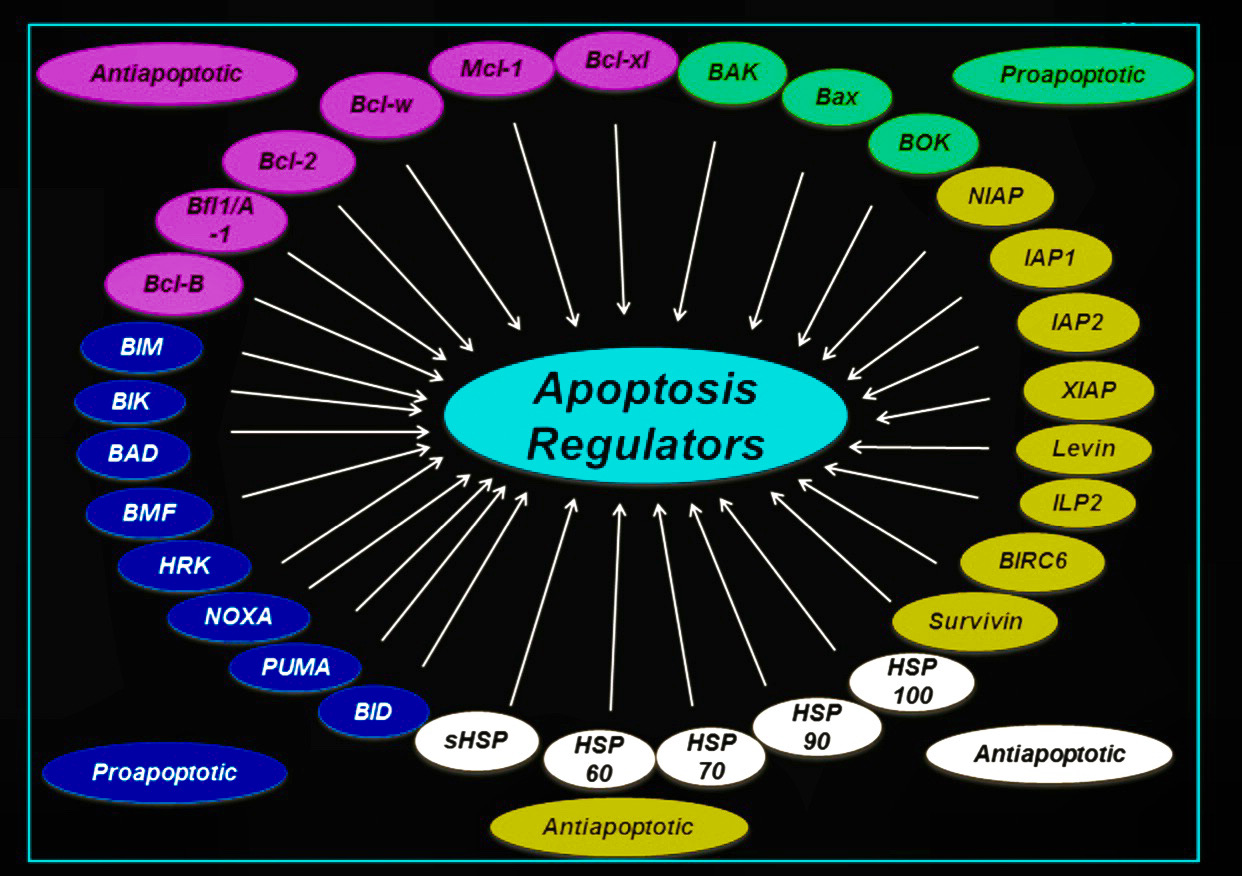
ATTENTION: THIS IS VERY IMPORTANT TO UNDERSTAND IF YOU WANT TO BECOME A ZOMBIE KILLING MACHINE
INSULIN IS ANTI-APOPTOTIC
WHICH MEANS IT
PROTECTS ZOMBIE CELLS FROM APOPTOSIS

THIS IS WHY YOU CAN’T KILL THAT STUBBORN FAT NO MATTER WHAT YOU SEEM TO DO. INSULIN IS BLOCKING YOU FROM doing so.
INGREDIENTS & Science

Acorus gramineus has been studied for its potential anti-aging properties, particularly its impact on senescence and related pathways. Research suggests it may modulate senolytic pathways such as PI3K/AKT, mTOR, and autophagy, targeting senescent cells to promote apoptosis. This contributes to reduced inflammation (via Nrf2, NF-kB) and enhanced cellular repair. By influencing these pathways, Acorus gramineus may support longevity and health span, though further research is needed to fully confirm these effects.

alantolactonE
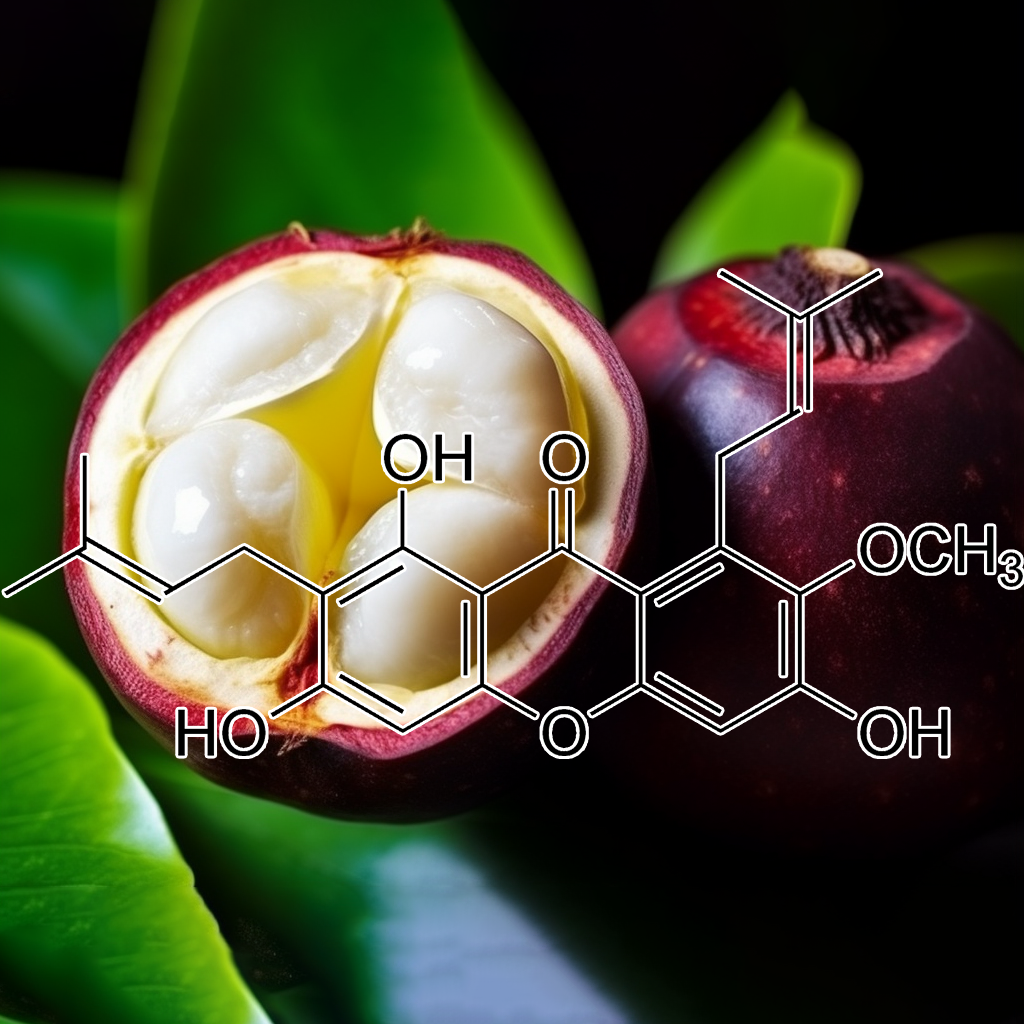
α-Mangostin
α-Mangostin, a xanthone derived from mangosteen, exhibits significant health benefits by targeting senescent cells and modulating key pathways such as PI3K/AKT/mTOR and TGF-β. It enhances cellular health through its anti-inflammatory and antioxidant properties, reducing senescence-associated secretory phenotype (SASP) factors. α-Mangostin promotes autophagy and apoptosis, providing protective effects against oxidative stress and potentially preventing vascular complications in diabetes. Its multifaceted action underscores its promise as a therapeutic agent for longevity and overall health enhancement.

Alpine Rose (Rhododendron ferrugineum L.)
Alpine Rose (Rhododendron ferrugineum L.) extract contains senolytic compounds like hyperoside and taxifolin, which target senescent cells and reduce the senescence-associated secretory phenotype (SASP). This extract promotes collagen production, enhances skin elasticity, and supports epidermal regeneration, offering significant anti-aging benefits. By activating cellular pathways involved in autophagy and apoptosis, Alpine Rose extract contributes to skin rejuvenation and overall health, making it a promising ingredient in anti-aging cosmetics.
Andrographolide, derived from *Andrographis paniculata*, is a bioactive compound known for its potential health benefits. It targets senolytic pathways, particularly PI3K/AKT and BCL-2, modulating autophagy and enhancing Nrf2 activity. Evidence indicates Andrographolide suppresses STAT3, promoting apoptosis and increasing sensitivity to cancer therapies like doxorubicin. Additionally, it inhibits MMP-9 expression via the NF-κB pathway, offering anti-inflammatory effects. These mechanisms suggest Andrographolide’s role in enhancing health and longevity by addressing cellular stress and inflammation.

Antrodia camphorata exhibits promising health benefits through its senolytic properties, targeting senescent cells and modulating critical pathways such as PI3K/AKT, mTOR, and autophagy. Its active compounds, like Antrocin, effectively inhibit the JAK2/STAT3 signaling pathway and induce apoptosis in cancer cells by enhancing Fas/APO-1 expression and suppressing NF-κB. Additionally, it influences key proteins, including Bcl-2 and Bax, potentially improving longevity and cellular stress responses, thereby supporting overall health and vitality.
APIGENIN
Apigenin, a natural flavonoid, effectively inhibits cancer cell growth by inducing G2/M arrest and modulating key proteins like cyclin B1 and CDK1. It also enhances telomerase activity and reduces cell senescence, particularly in dental pulp cells. By influencing pathways related to senescence, including autophagy and apoptosis, apigenin promotes an undifferentiated state, potentially targeting senescent cells. These mechanisms suggest apigenin’s role in improving health and longevity by mitigating the effects of cellular aging.
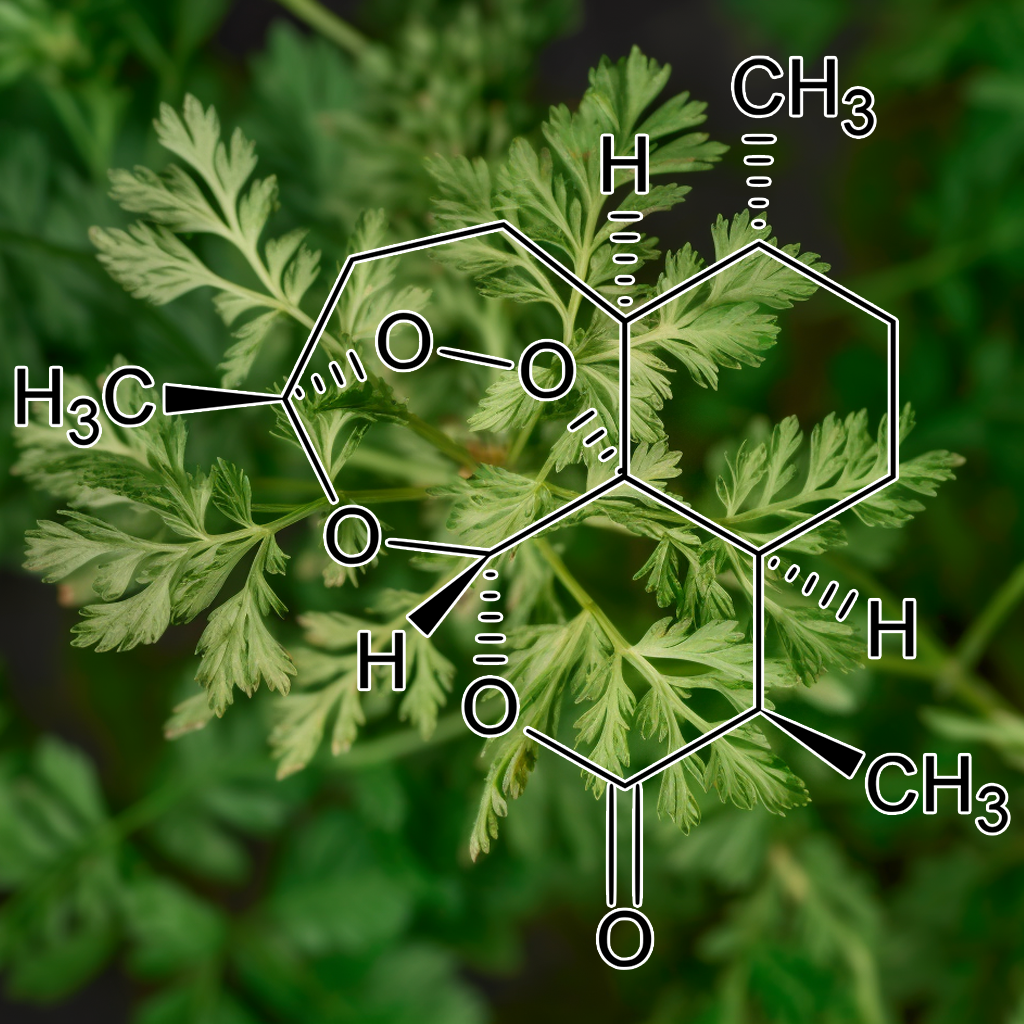
Artemisinin
Artemisinin, derived from Artemisia annua, is known for its selective induction of apoptosis in cancer cells and potential senolytic effects. It influences key pathways like SCAPs and PI3K/AKT, while modulating proteins such as Bcl-2 and Bax. By reducing senescence-associated secretory phenotype (SASP) factors and enhancing autophagy, Artemisinin promotes cellular resilience, which may contribute to improved longevity and health outcomes. Its capacity to induce cell cycle arrest in neuroblastoma further underscores its potential as an effective cancer treatment.

Artocarpin, a natural compound, demonstrates significant senolytic effects by targeting senescent cells and modulating pathways such as PI3K/AKT, BCL-2, and mTOR. It enhances autophagy and reduces the senescence-associated secretory phenotype (SASP), promoting cellular resilience and longevity. Additionally, artocarpin induces apoptosis in non-small cell lung cancer (NSCLC) by generating reactive oxygen species (ROS) and activating the p53-independent ROS/NF-kB/c-Myc/Noxa pathway, highlighting its potential as an effective anticancer agent.

Aspalathin, a flavonoid found in rooibos tea, exhibits potential health benefits through its antioxidant properties. It enhances Nrf2 expression, protecting cardiomyocytes from oxidative stress and apoptosis in diabetic conditions. While direct evidence linking aspalathin to senolytic pathways is limited, its influence on pathways like PI3K/AKT and mTOR suggests a role in targeting senescent cells and the senescence-associated secretory phenotype (SASP), which may contribute to improved longevity and overall health.

Astragalus complanatus seed extract
Astragalus demonstrates promising senolytic properties by modulating key signaling pathways, including PI3K/AKT and BCL-2, which target senescent cells. By reducing senescence-associated secretory phenotype (SASP) factors, it may promote autophagy and enhance longevity. Additionally, the induction of apoptosis through regulators like Bax and the modulation of cyclins and CDKs further supports its anti-aging benefits. These effects collectively contribute to improved health and longevity, making Astragalus a potential candidate in age-related therapies.


Bavachin, a phytoestrogen from Psoralea corylifolia, exhibits potential as a senolytic agent by targeting senescent cells and modulating key pathways like PI3K/AKT, BCL-2, and mTOR. It induces apoptosis in multiple myeloma cells by inhibiting NF-κB and STAT3 while activating caspases. By reducing senescence-associated secretory phenotype (SASP) factors, bavachin may enhance cellular health and longevity, potentially mitigating age-related diseases and promoting overall resilience. Its promising effects make it a candidate for future cancer therapies.
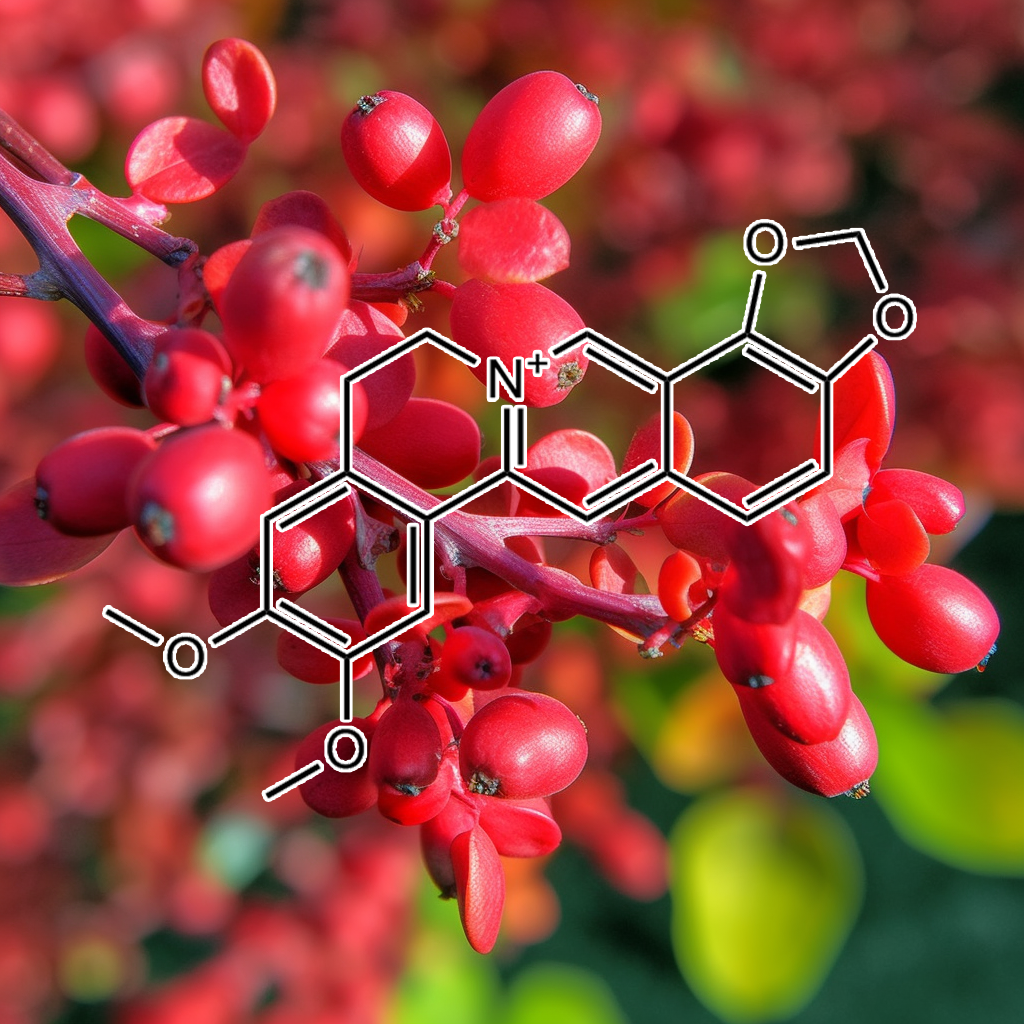
Berberine, derived from Coptis chinensis, exhibits significant anti-aging properties by targeting senescent cells and modulating key senolytic pathways including PI3K/AKT, BCL-2, and mTOR. It enhances longevity and cellular health by promoting apoptosis, regulating the inflammatory response, and activating autophagy. Additionally, Berberine’s antioxidant effects and ability to suppress SASP support its potential as a natural remedy for aging-related diseases. This evidence-based approach underscores Berberine’s role in improving healthspan and mitigating age-associated inflammation.


Betulinic acid demonstrates senolytic properties by targeting senescent cells and modulating vital cellular pathways such as PI3K/AKT and mTOR. It effectively decreases pro-survival proteins like Bcl-2 and cyclin D1 while increasing pro-apoptotic markers such as Bax. These actions reduce the senescence-associated secretory phenotype (SASP) and enhance autophagy, promoting cellular health and longevity. This therapeutic potential may support age-related health conditions and exhibit anticancer effects, highlighting its significance in modern health research.

Brevilin A, a sesquiterpene lactone from Centipeda minima, exhibits senolytic properties by targeting senescent cells and modulating critical pathways such as PI3K/AKT/mTOR and JAK-STAT. By reducing the senescence-associated secretory phenotype (SASP), it promotes apoptosis and autophagy, enhancing health and longevity. Its effects on proteins like Bcl-2, Bax, and Survivin contribute to improved cellular outcomes, potentially mitigating age-related diseases and promoting overall well-being through targeted cellular rejuvenation.
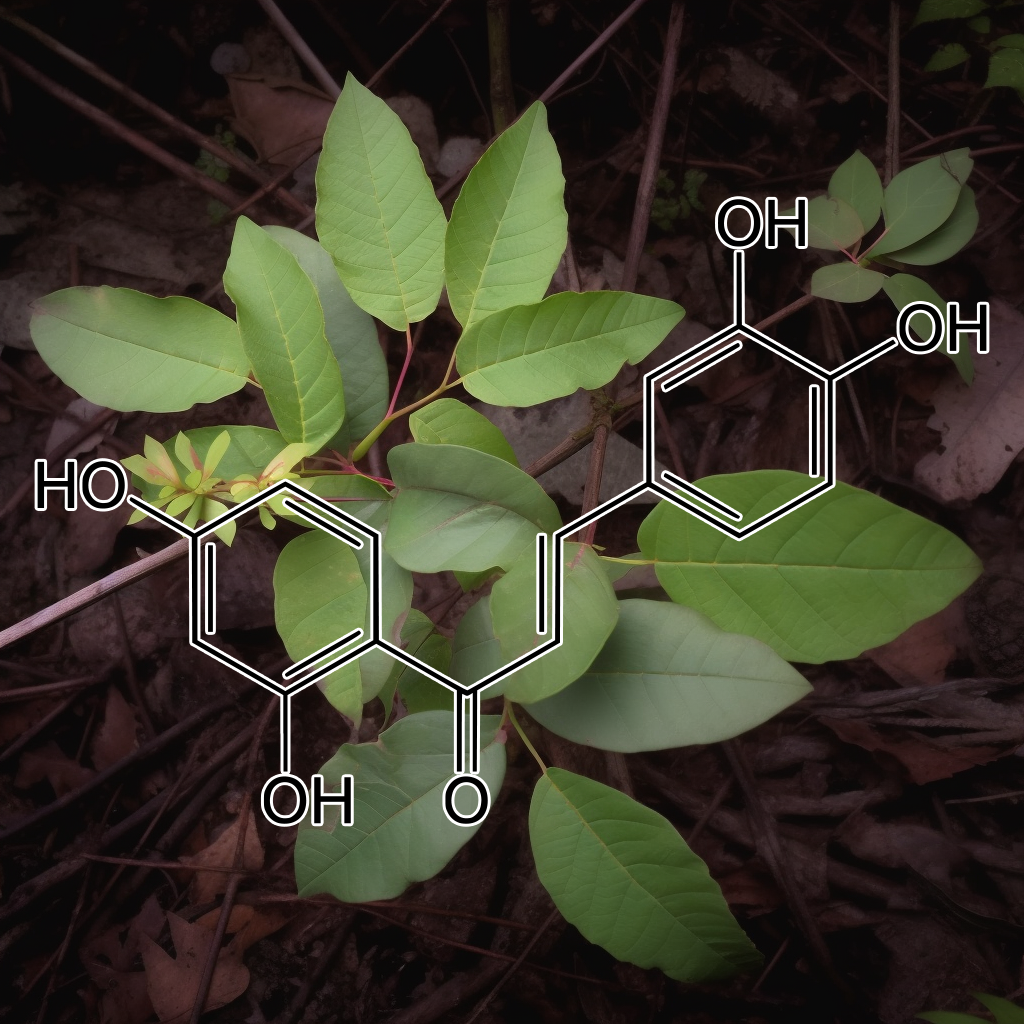
Butein, a senolytic compound, effectively targets senescent cells by modulating key pathways like PI3K/AKT, mTOR, and BCL-2, enhancing autophagy and influencing SASP factors. Its ability to inhibit NF-κB activation sensitizes cells to TRAIL-induced apoptosis, offering potential therapeutic benefits in cancer treatment. By degrading Hsp90 client proteins, butein combats drug-resistant cancer cells, promoting health and longevity through cellular rejuvenation and the mitigation of aging-related diseases.
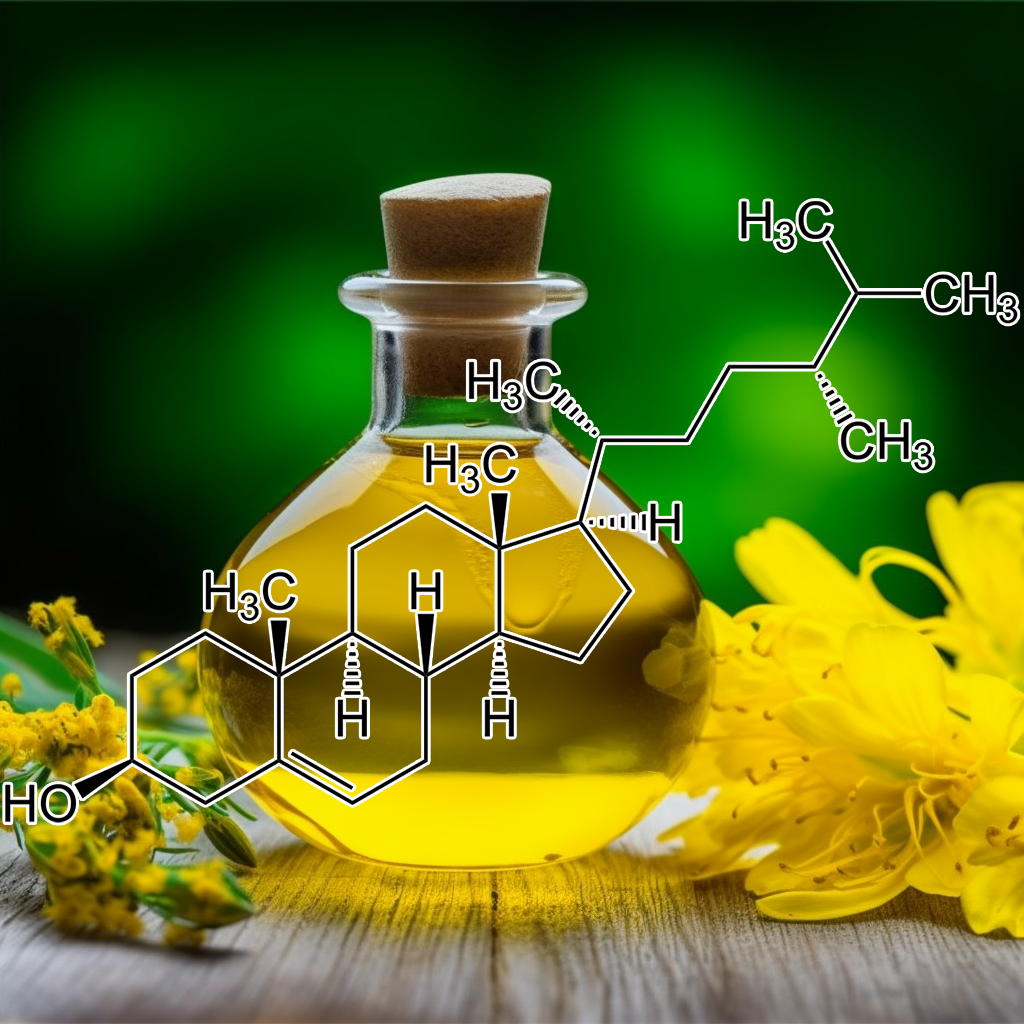
Campesterol, a phytosterol, may influence cellular aging by modulating senolytic pathways, including the PI3K/AKT and BCL-2 signaling pathways. This compound has been shown to down-regulate anti-apoptotic proteins like BCL-2 and BCL-xL while up-regulating pro-apoptotic factors such as Bax and Bad, potentially enhancing apoptosis in senescent cells. Additionally, its interaction with autophagy and Nrf2 signaling may support healthspan and longevity by reducing inflammation associated with the senescence-associated secretory phenotype (SASP).

Carnosic acid, derived from rosemary, shows promise in enhancing cellular health and longevity by targeting senescent cells. It modulates critical senolytic pathways, including PI3K/AKT and autophagy, while influencing apoptosis regulators like Bcl-2 and c-FLIP. Research indicates that carnosic acid promotes TRAIL-mediated apoptosis in renal carcinoma cells through the up-regulation of DR5, Bim, and PUMA. These mechanisms collectively support the reduction of senescent cell burden, potentially improving healthspan and longevity.

casticin
Casticin, a natural compound, shows promise in cancer treatment by inducing apoptosis through the downregulation of survival proteins like Bcl-2 and XIAP. Its ability to enhance TRAIL-induced apoptosis in colon cancer cells suggests potential applications in targeting senescent cells and modulating the senescence-associated secretory phenotype (SASP). By generating reactive oxygen species, Casticin may influence senolytic pathways, contributing to improved health and longevity by eliminating dysfunctional cells and promoting cellular resilience.
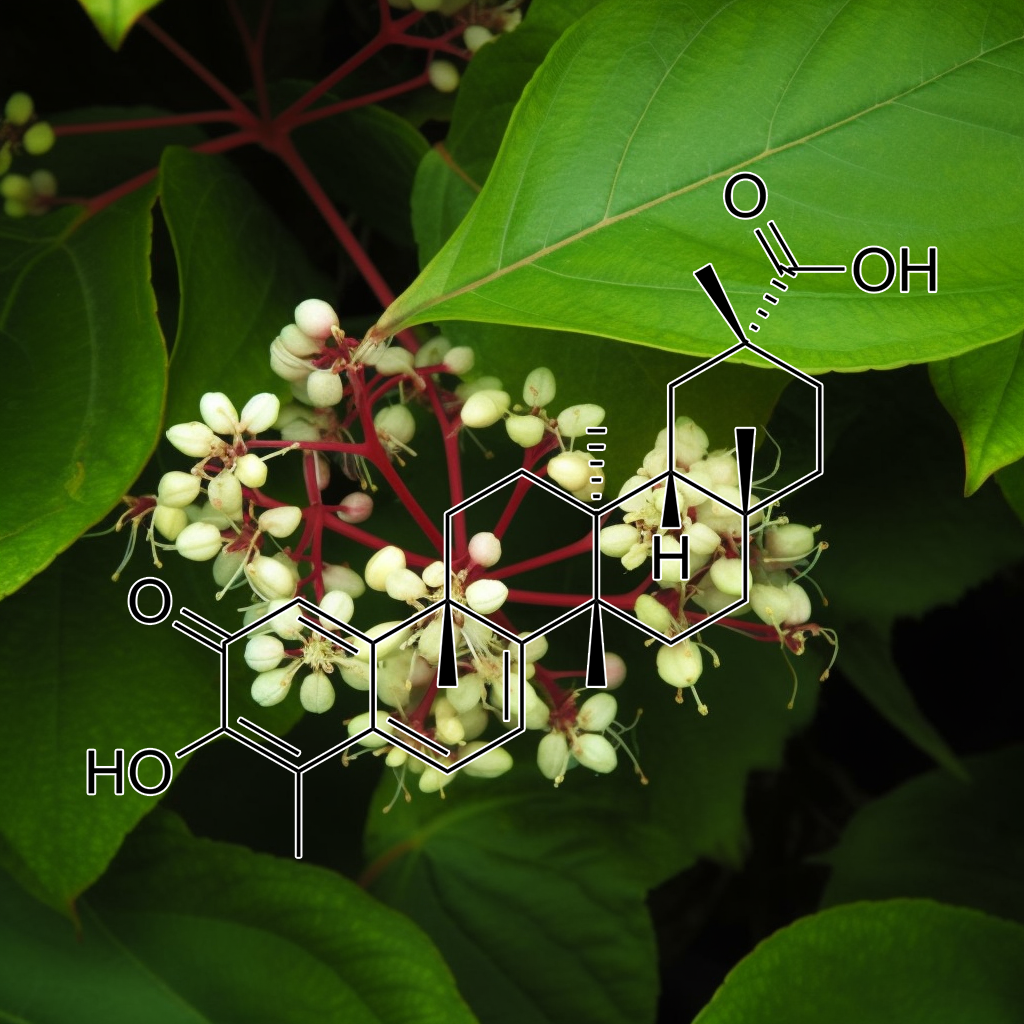
Celastrol, a natural compound, demonstrates significant anti-senescence effects by targeting senescent cells through pathways like PI3K/AKT and mTOR. It modulates the senescence-associated secretory phenotype (SASP), enhances autophagy, and influences key proteins such as BAX and BCL-2. Additionally, Celastrol reduces oxidative stress and promotes apoptosis, showing promise in treating age-related diseases and cancer. Its ability to improve vascular health further supports its therapeutic potential, enhancing longevity and overall well-being.

CHICORIC ACID
Chicoric acid shows promise in promoting health and longevity by modulating senolytic pathways. It targets key pathways such as PI3K/AKT, mTOR, and Nrf2, influencing the dynamics of senescent cells and the senescence-associated secretory phenotype (SASP). By inducing apoptosis in 3T3-L1 preadipocytes through mitochondrial mechanisms and generating reactive oxygen species, chicoric acid may help mitigate obesity-related effects. Its interactions with proteins like BAX, BIM, and HSP70 underscore its potential in enhancing cellular health.
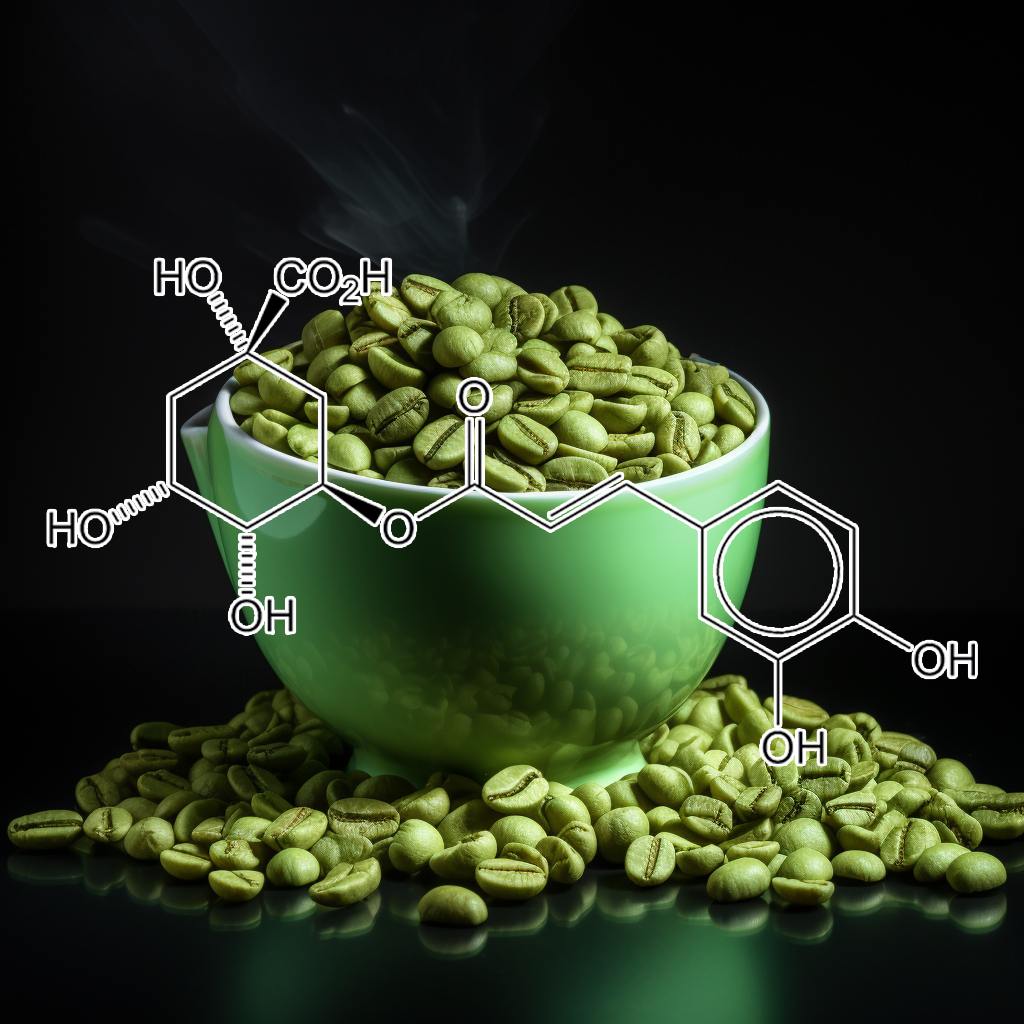
Chlorogenic acid, a bioactive compound from Euonymus alatus, influences senescence and longevity through modulation of key pathways such as PI3K/AKT, Nrf2, autophagy, and apoptosis-related proteins like Bcl-2 and Bcl-xl. Its ability to inhibit MMP-9 and reduce HMGB1 release highlights its potential in cancer chemoprevention and immune response enhancement. By targeting senescent cells and mitigating the senescence-associated secretory phenotype (SASP), chlorogenic acid promotes healthspan and longevity through antioxidant and anti-inflammatory effects.

Cianidanol, derived from Centipeda minima, shows promise as a senolytic compound by targeting senescent cells. It influences critical pathways such as PI3K/AKT and mTOR, enhancing apoptosis through modulation of BCL-2 family proteins like BAX and BAK. By reducing the senescence-associated secretory phenotype (SASP) and promoting autophagy, Cianidanol may contribute to improved cellular health and longevity, potentially preventing age-related diseases. Further investigation into natural senolytics is essential for advancing anti-aging therapies.

Costunolide, a natural sesquiterpene lactone, exhibits promising senolytic properties by targeting critical pathways such as PI3K/AKT and BCL-2. This compound promotes the clearance of senescent cells, influencing the senescence-associated secretory phenotype (SASP), apoptosis, and autophagy. Its ability to induce apoptosis in platinum-resistant cancer cells underscores its potential in enhancing cellular resilience and longevity. Overall, costunolide may serve as a vital agent in advancing health by mitigating the effects of cellular aging.

Curcumin demonstrates significant health benefits by targeting senescent cells and modulating key pathways, including PI3K/AKT, BCL-2, and Nrf2. Research indicates it can alleviate cognitive dysfunction and oxidative stress in aging models, such as d-galactose-treated mice, and prevent mitochondrial dysfunction in senescence-accelerated mice. By enhancing autophagy and influencing apoptosis-related proteins, curcumin promotes cellular health and longevity, showcasing its therapeutic potential against neurodegenerative diseases like Alzheimer’s.

Cucurbitacin D exhibits potent senolytic effects by targeting senescent cells and modulating key pathways like PI3K/AKT, mTOR, and BCL-2, promoting apoptosis and autophagy. It influences SASP, cGAS-STING, and Nrf2, contributing to reduced inflammation and enhanced cellular health. By regulating proteins like BAX, BCL-xl, and caspase-3, Cucurbitacin D supports longevity and combats age-related decline. Its role in targeting TLR4, STAT3, and NF-κB further underscores its potential for promoting health and lifespan extension through senescence modulation.

Cycloastragenol
Cycloastragenol, a potent compound derived from Astragalus, supports longevity by activating telomerase and protecting against cellular senescence. Its potential senolytic activity targets senescent cells, modulating pathways like PI3K/AKT, mTOR, and Nrf2. By reducing SASP factors, Cycloastragenol may help inhibit inflammation and oxidative stress. It interacts with apoptosis-regulating proteins (BCL-2, BAX) and promotes autophagy. These combined effects enhance cellular health, reduce aging-related damage, and contribute to overall vitality, offering promising benefits for healthy aging and longevity.

Daidzein, a powerful soy isoflavone, plays a key role in tackling cellular aging by targeting senescent cells and unlocking senolytic pathways. It influences the PI3K/AKT and mTOR cascades, reducing the harmful SASP, and activates autophagy to clear out damaged cells. By modulating key apoptosis proteins like BCL-2 and Bax, and interacting with pathways like Nrf2 and NF-kB, daidzein combats inflammation and oxidative stress, promoting longevity and cellular rejuvenation for healthier aging.

Decursin, a powerful compound from Angelica gigas, emerges as a potent force in anti-aging research. It targets senescent cells and dampens the harmful SASP, while engaging key senolytic pathways like PI3K/AKT and BCL-2. By balancing autophagy and triggering apoptosis through BAX, PUMA, and CASPASE-3, decursin helps clear damaged cells. Its ability to reduce inflammation via TLR4, NF-κB, and STAT3 offers new hope for enhancing longevity, making it a fascinating prospect in the fight against aging.

Targeting cellular senescence through key pathways like PI3K/AKT, mTOR, Nrf2, and autophagy, delphinidin plays a significant role in reducing the effects of SASP (Senescence-Associated Secretory Phenotype) and regulating apoptotic proteins such as BCL-2, BAX, and CASPASE 3. Its strong anti-inflammatory, antioxidant, and senolytic properties help combat age-related cellular damage, promoting longevity and supporting healthier aging by addressing the underlying mechanisms of cellular senescence.

Cardiac glycosides, found in plants like Digitalis purpurea, may influence cellular senescence by modulating pathways such as PI3K/AKT, autophagy, and apoptosis regulators like Bcl-2. While definitive links to senolytic activity remain under investigation, these compounds show potential in affecting cellular health, autophagy, and longevity. Their role in regulating senescent cell pathways, including mTOR and Nrf2, offers a promising but unconfirmed avenue for enhancing healthspan and lifespan through targeting aging-related processes.

Diosmin, a bioflavonoid, shows potential in targeting senescent cells and mitigating inflammation linked to aging. It may influence key pathways like PI3K/AKT, mTOR, and NF-κB, which are associated with cellular senescence, SASP (senescence-associated secretory phenotype), and autophagy. Diosmin’s ability to reduce oxidative stress by modulating Nrf2 and supporting apoptosis regulation, through proteins like Bcl-2 and BAX, suggests it could play a role in enhancing longevity and healthspan. Further studies are ongoing to confirm these effects.


HOELEN (Poria Cocos)
Hoelen (Poria cocos) exhibits potential senolytic properties by targeting senescent cells and influencing key pathways like PI3K/AKT, mTOR, and autophagy. It modulates apoptosis regulators like Bcl-2, BAX, and caspase-3, while impacting stress-related proteins such as HSP70 and HSP90. Additionally, Poria cocos may influence the Nrf2 and NF-κB pathways, contributing to reduced inflammation and improved cellular health. These actions support longevity by reducing the harmful effects of senescent cells and promoting healthy aging.

EF24, a curcumin analog derived from turmeric root, has shown potential as a senolytic agent, targeting senescent cells and related pathways, including PI3K/AKT, Bcl-2, Nrf2, and mTOR. It promotes autophagy, regulates apoptosis through proteins like Bcl-xL and Bax, and modulates inflammation via NF-kB. EF24 also interacts with heat shock proteins (HSPs) and influences key regulators like STAT3 and caspases, supporting healthy aging and longevity by mitigating the harmful effects of senescent cells.

Epigallocatechin gallate (EGCG) is a potent polyphenol with senolytic effects, promoting the clearance of senescent cells. EGCG influences key pathways such as PI3K/AKT, BCL-2, Nrf2, mTOR, and autophagy, reducing the harmful effects of the senescence-associated secretory phenotype (SASP). It also modulates apoptosis-related proteins (BAX, BCL-XL) and immune responses via the cGAS-STING and TLR4 pathways. EGCG’s impact on these mechanisms contributes to enhanced longevity, reduced inflammation, and better cellular health.

Ellagic acid
Ellagic acid has been linked to senescence and longevity pathways, targeting senescent cells by modulating apoptosis, autophagy, and senolytic pathways like PI3K/AKT, BCL-2, and mTOR. It influences key regulators like Nrf2, cGAS-STING, STAT3, and NF-kB, promoting cellular health and reducing SASP (Senescence-Associated Secretory Phenotype). Its role in enhancing autophagy and apoptosis markers such as CASPASE-3 and BAX contributes to its potential in improving longevity and health outcomes, offering a promising avenue for age-related therapies.

Embelin, a natural quinone compound, exhibits senolytic activity by targeting senescent cells and modulating key survival pathways, including BCL-2, PI3K/AKT, and mTOR. It promotes apoptosis in senescent cells, inhibits SASP, and influences autophagy. Embelin also interacts with apoptosis regulators like Bcl-xl, Bax, and IAPs, and affects cGAS-STING and Nrf2 pathways. This positions embelin as a potential therapeutic agent in enhancing healthspan, longevity, and age-related disease management by eliminating harmful senescent cells.

Emodin

Eriodictyol
Eriodictyol, a flavonoid, shows potential in targeting cellular senescence by modulating key pathways like PI3K/AKT, mTOR, and Nrf2. It may impact senolytic activity, promoting autophagy, apoptosis, and reducing SASP (senescence-associated secretory phenotype). By influencing proteins such as Bcl-2, BAX, and caspases, it could support healthy aging and longevity. Eriodictyol’s effects on senescent cells and pathways like STAT3, NF-κB, and cGAS-STING offer promising anti-aging benefits, enhancing cellular repair and survival mechanisms.
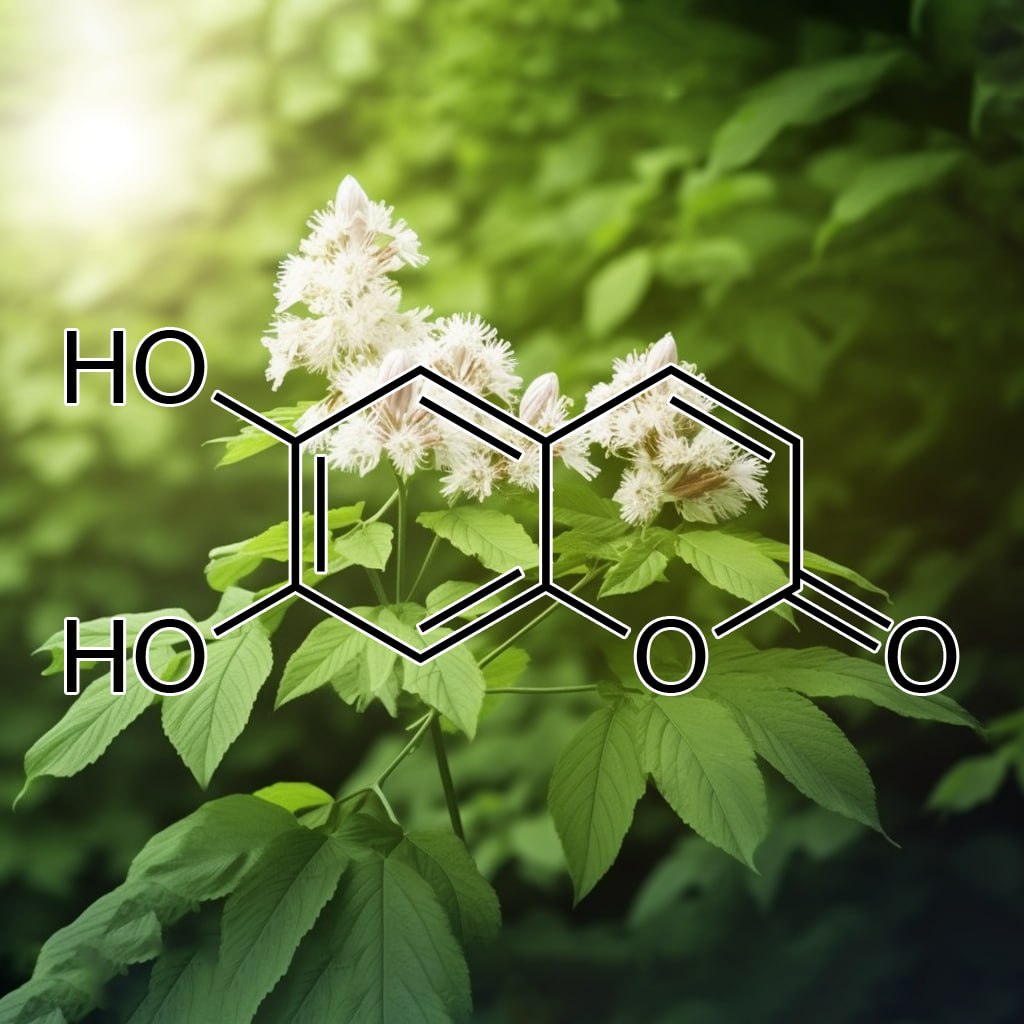
Esculetin
Esculetin, a natural coumarin compound, shows potential in targeting senescent cells through senolytic pathways. It modulates key mechanisms like PI3K/AKT, BCL-2 family proteins, apoptosis, and autophagy, which play roles in cellular senescence. Esculetin may inhibit SASP, reduce inflammation via Nrf2 and NF-κB, and activate caspase-3 to promote apoptosis in damaged cells. These actions contribute to improved healthspan and longevity, positioning esculetin as a promising agent in anti-aging and cellular rejuvenation research.
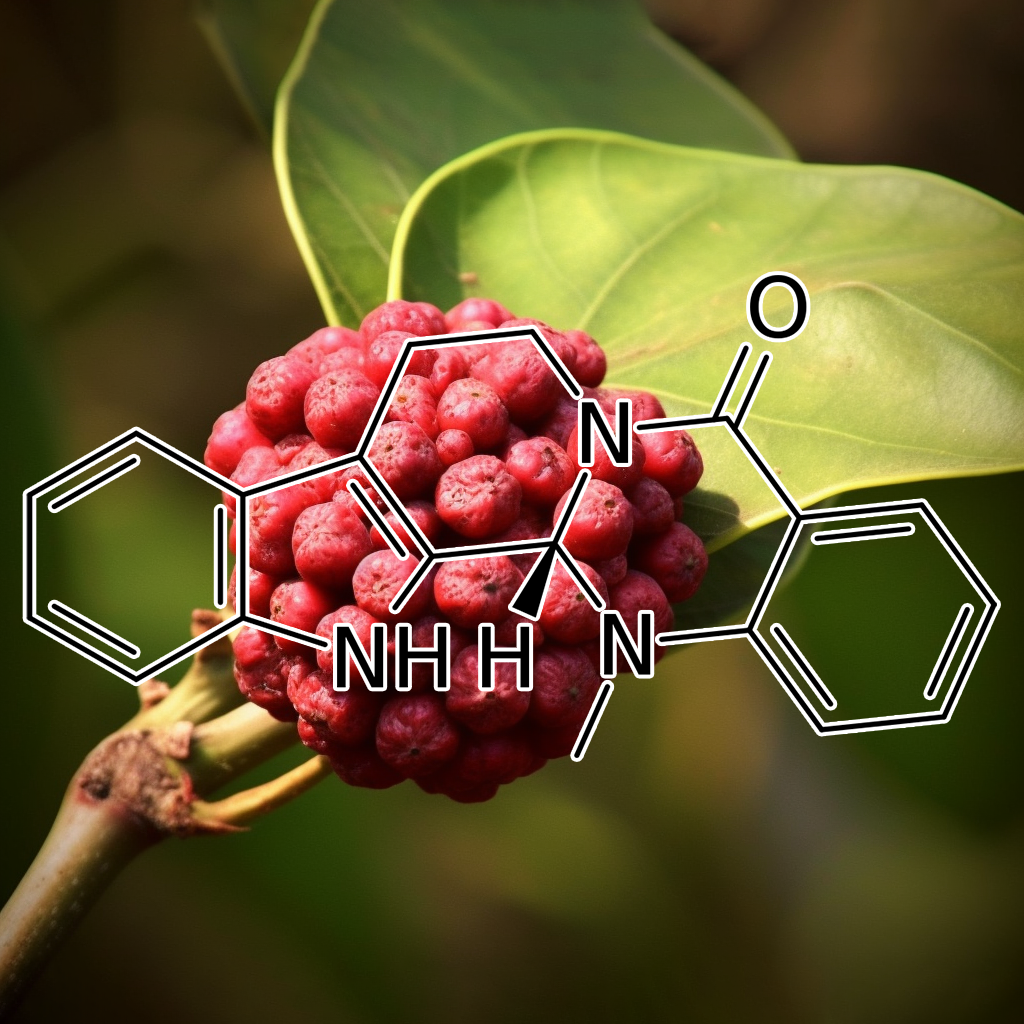
Evodiamine, a bioactive compound, exhibits potential senolytic activity by targeting senescent cells. It interacts with pathways like PI3K/AKT, mTOR, and Bcl-2 family proteins, influencing apoptosis and autophagy. Additionally, evodiamine may modulate the Nrf2 and NF-kB pathways, promoting cellular resilience and reducing SASP (senescence-associated secretory phenotype). These mechanisms contribute to cellular rejuvenation, with implications for enhanced longevity, reduced inflammation, and improved healthspan, making evodiamine a promising candidate in anti-aging and senescence-targeted therapies.

A natural flavonoid, fisetin, acts as a potent senolytic compound, effectively targeting senescent cells and reducing SASP (senescence-associated secretory phenotype). It regulates vital pathways like PI3K/AKT, mTOR, and BCL-2 family proteins, promoting autophagy, apoptosis, and reducing inflammation. By influencing the Nrf2 and cGAS-STING pathways, fisetin enhances cellular repair, minimizes oxidative stress, and supports longevity. Its ability to clear senescent cells positions it as a promising therapeutic for age-related diseases and healthy aging.
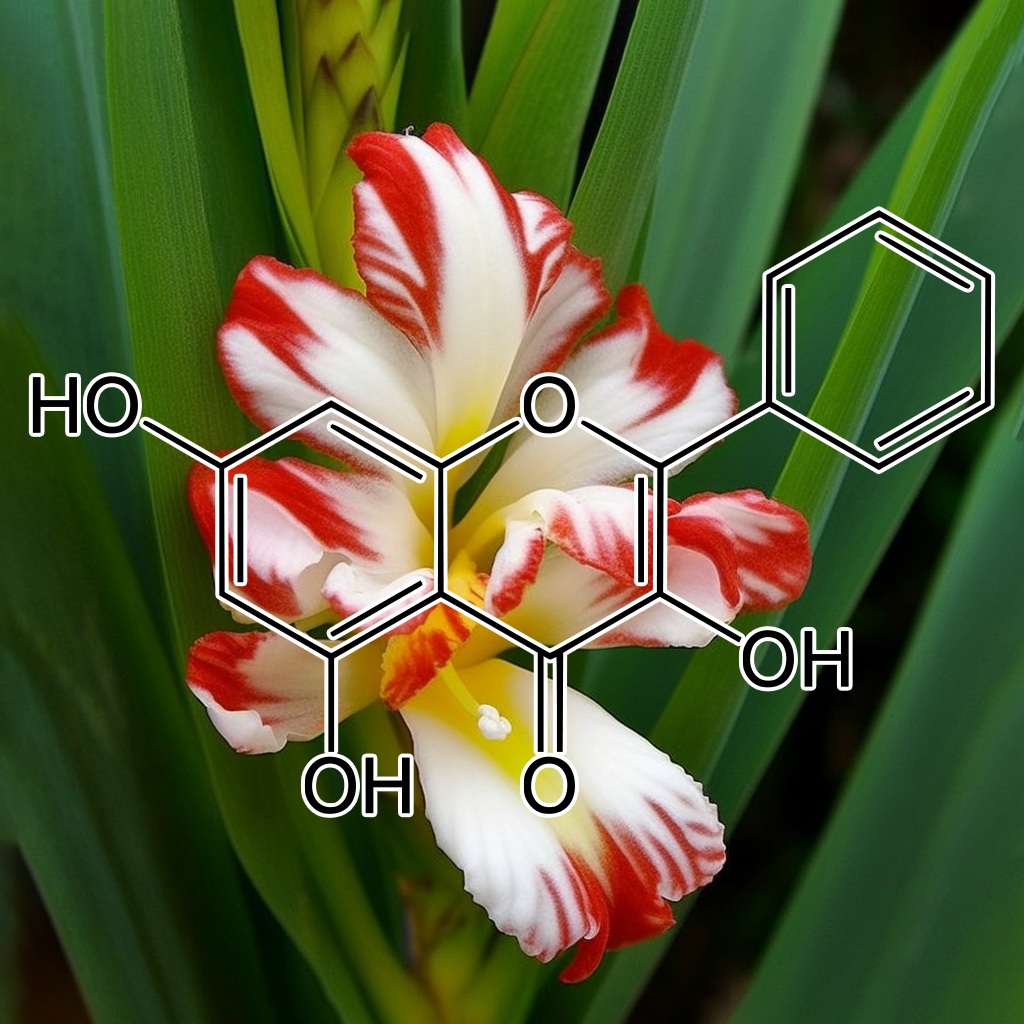
Galangin (Alpinia officinarum)
Targeting senescent cells and key pathways like PI3K/AKT, BCL-2, and mTOR, galangin has shown promise in enhancing longevity. It reduces the harmful effects of SASP (senescence-associated secretory phenotype) while promoting autophagy, apoptosis, and cellular repair. By modulating Nrf2, cGAS-STING, and STAT3, it helps protect against oxidative stress and inflammation, offering potential benefits for healthy aging and longevity, making it an emerging compound for age-related health optimization.
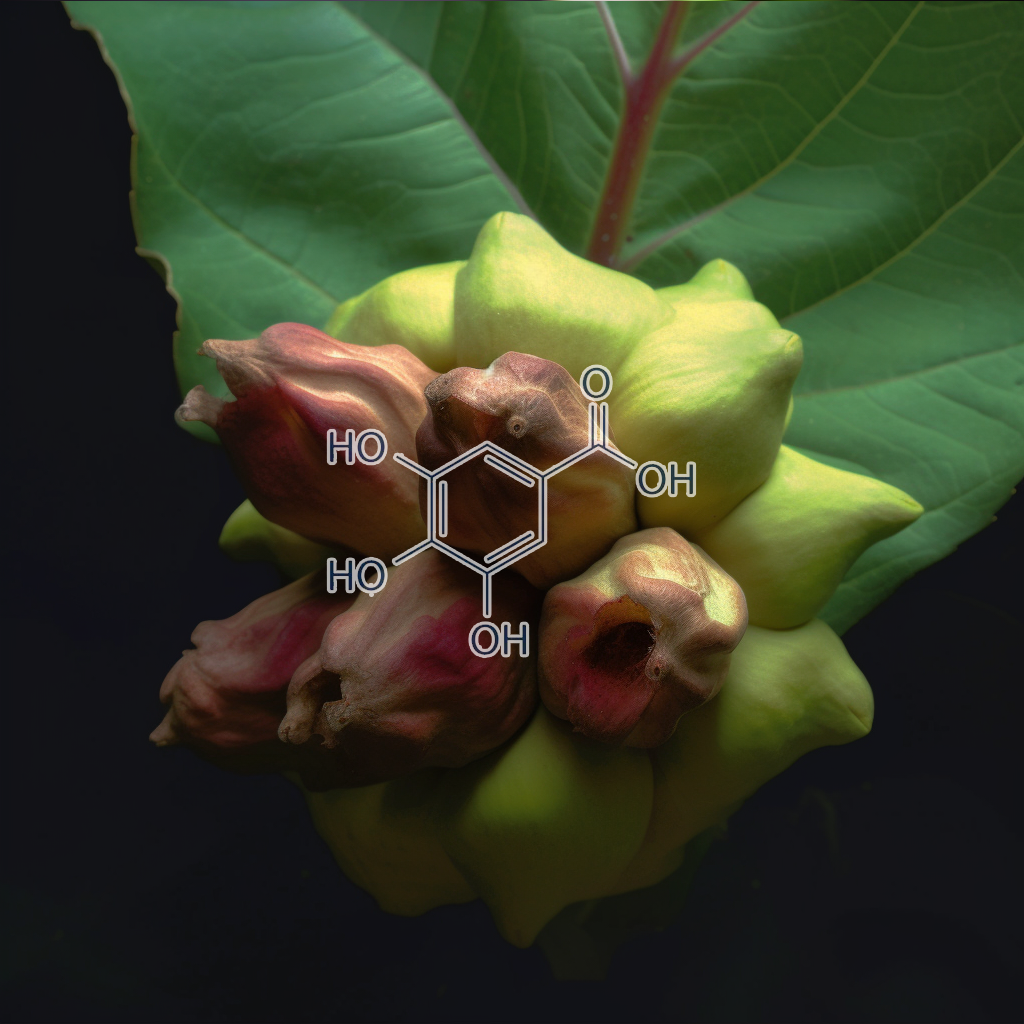
Gallic acid, a polyphenol, has potential senolytic effects by targeting senescent cells and modulating key pathways like PI3K/AKT, Bcl-2, mTOR, and Nrf2. It influences apoptosis, autophagy, and inflammation, potentially reducing the harmful SASP (Senescence-Associated Secretory Phenotype). By regulating Bcl-xl, Bax, PUMA, and caspases, it promotes cellular health. Its interaction with pathways like STAT3, NF-κB, and cGAS-STING may enhance longevity, making it a promising compound in aging and healthspan research.
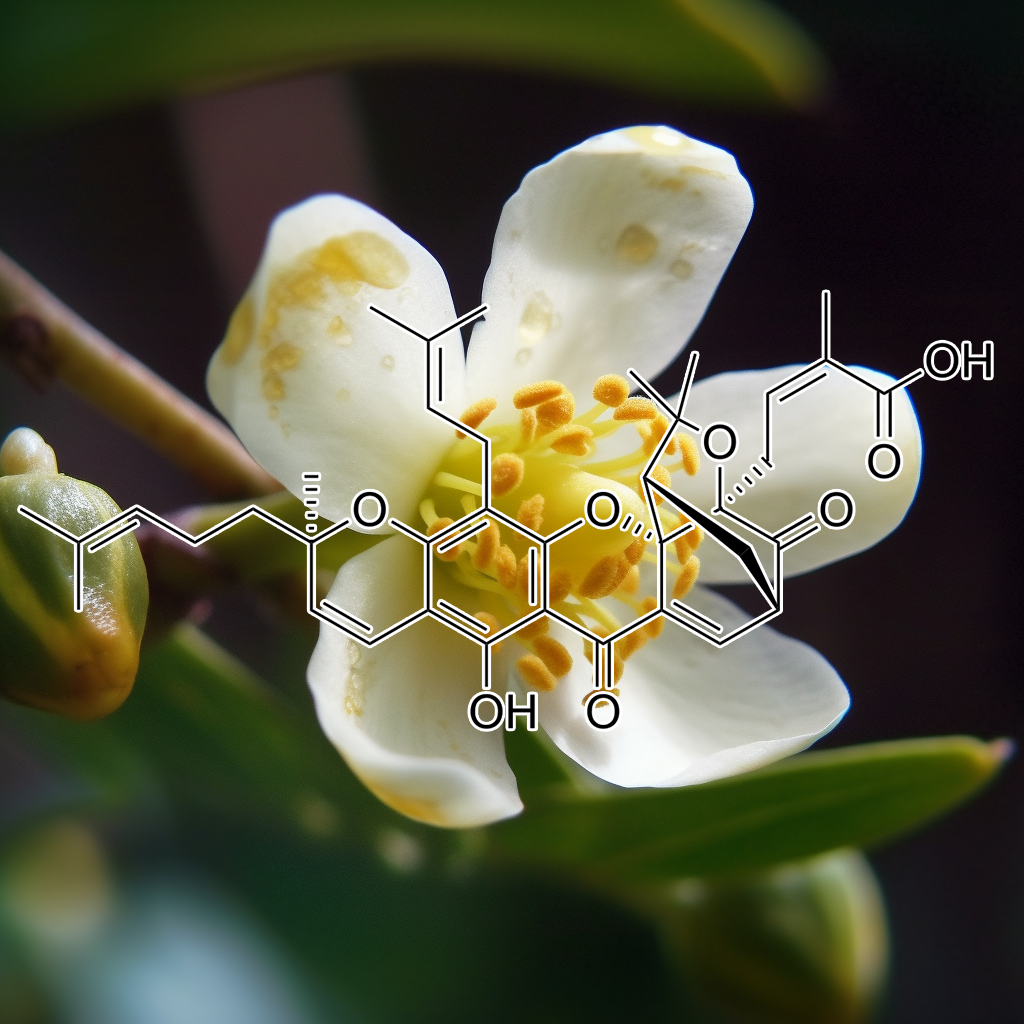
Gambogic Acid has shown potential as a senolytic agent, selectively targeting senescent cells to improve health and longevity. It influences key pathways such as PI3K/AKT, BCL-2, mTOR, autophagy, and apoptosis regulators like BAX, Bcl-xL, and caspase-3. By modulating these pathways, Gambogic Acid may help reduce SASP factors, promote apoptosis, and support cellular rejuvenation, contributing to anti-aging effects. Its interaction with senolytic pathways suggests promise for improving lifespan and overall health.

Ganoderma lucidum (Reishi) is linked to anti-aging by targeting cellular senescence and associated pathways. It supports apoptosis, autophagy, and modulates senolytic pathways, including PI3K/AKT, mTOR, and Nrf2. Reishi impacts SASP, reducing inflammation and oxidative stress, while enhancing longevity by modulating proteins like BCL-2, BAX, and caspase-3. Its effects on TLR4, NF-kB, and STAT3 contribute to improved immune response and healthspan. Reishi’s unique properties make it a valuable candidate for age-related health optimization.
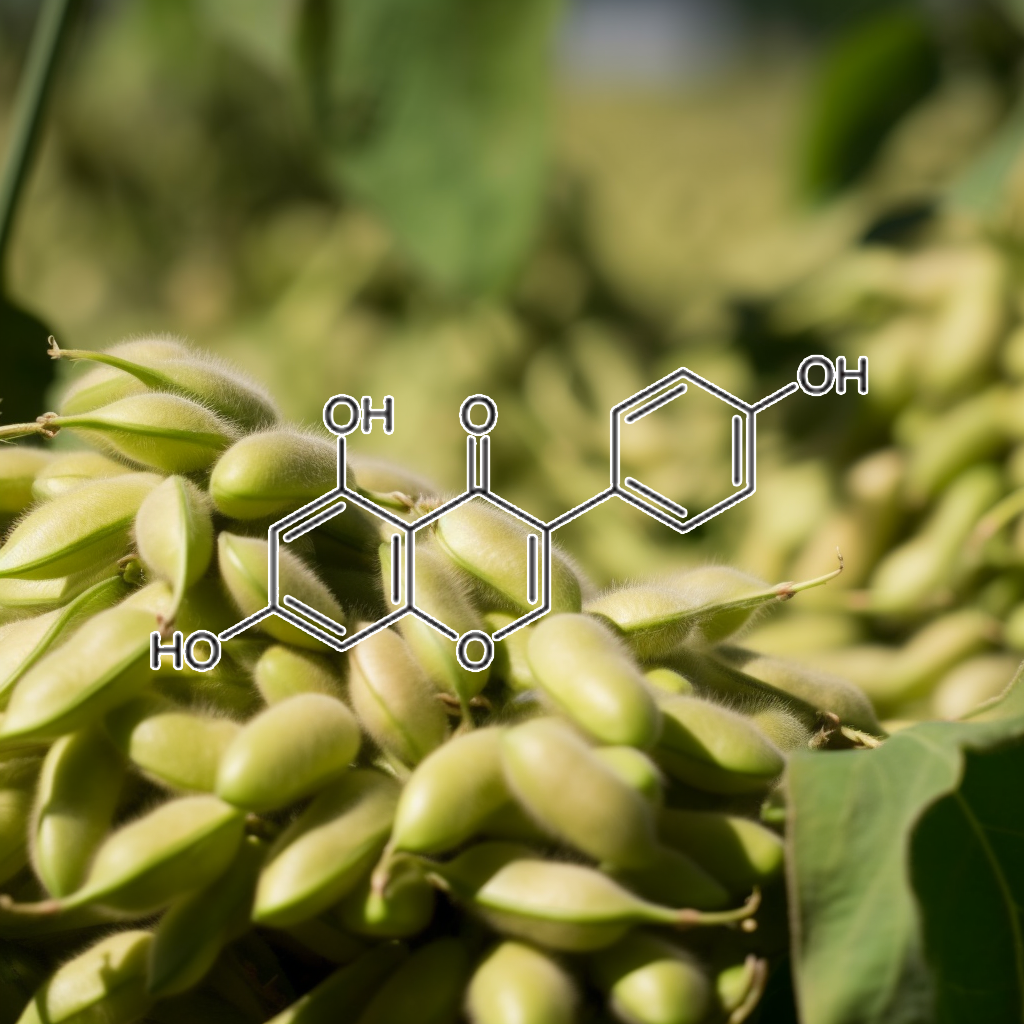
Genistein, a natural isoflavone, has been shown to act on multiple pathways related to senescence and longevity. It targets senescent cells by modulating pathways like PI3K/AKT, mTOR, and Nrf2, promoting autophagy and apoptosis. Genistein’s influence on SASP, BCL-2 family proteins, and caspase-3 suggests its role as a senolytic agent. It may also impact immune responses via the cGAS-STING and NFKB pathways, supporting healthy aging by reducing inflammation and enhancing cellular repair processes.
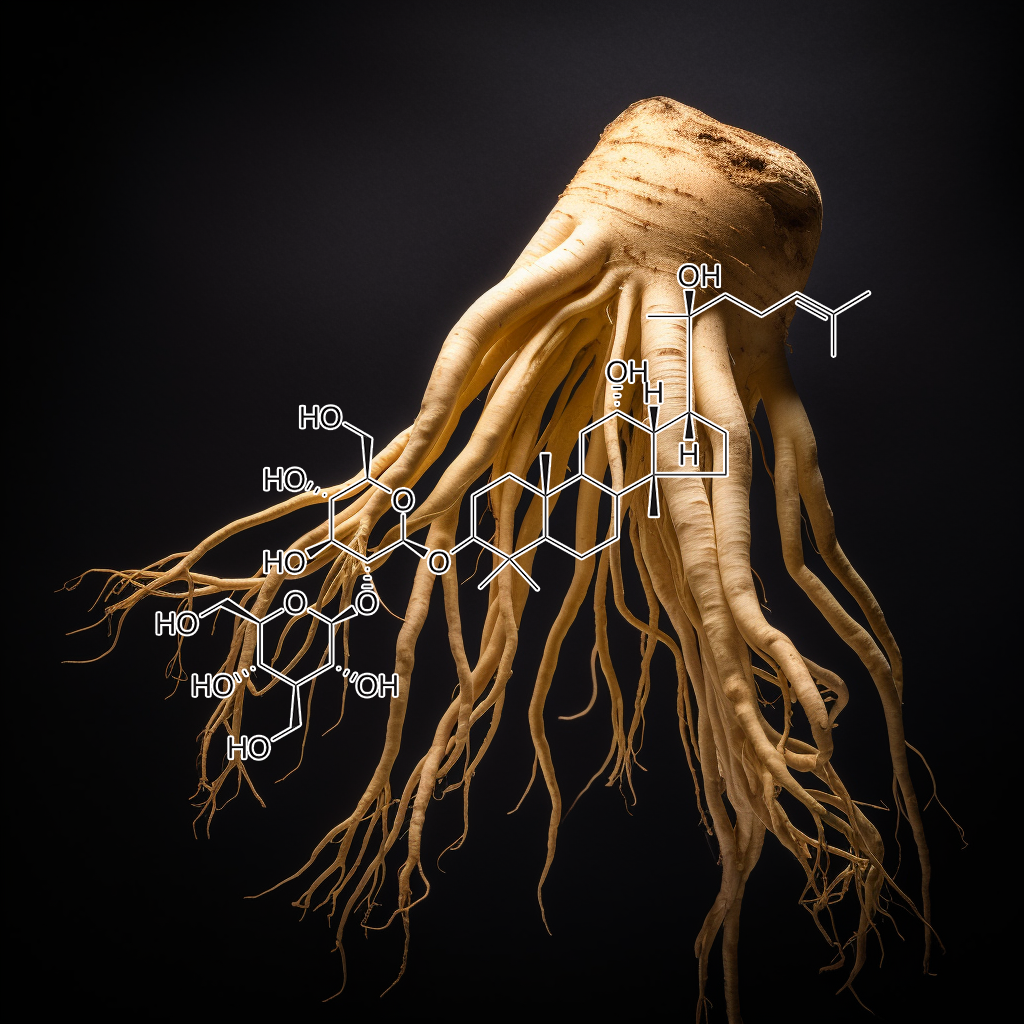
Ginsenoside Rg3, a bioactive compound in Panax ginseng, exhibits potential senolytic properties by targeting senescent cells and modulating pathways like PI3K/AKT, mTOR, and Bcl-2. It may enhance autophagy, apoptosis, and reduce inflammation by inhibiting SASP factors, TLR4, and STAT3/NF-kB pathways. Evidence suggests Rg3 contributes to longevity and health by promoting cellular homeostasis, reducing senescence, and activating apoptosis-related proteins like Caspase-3, thus supporting its role in anti-aging therapies.

Glycitein, a soy isoflavone, holds promise in the battle against aging by targeting senescent cells. It influences pathways like PI3K/AKT, Bcl-2, and mTOR, helping clear damaged cells, reduce inflammation (SASP), and boost cellular cleanup (autophagy). By activating stress defenses through Nrf2 and cGAS-STING, glycitein supports healthy aging, promoting longevity. Its ability to fine-tune apoptosis and inflammation control may offer protection against age-related diseases, enhancing vitality as we grow older.

Gossypol is emerging as a promising senolytic agent, targeting senescent cells to reduce the senescence-associated secretory phenotype (SASP) and enhance health and longevity. By inhibiting Bcl-2 and Bcl-xL, gossypol induces apoptosis and promotes autophagy, effectively disrupting cancer pathways and supporting cellular rejuvenation. Its dual role as an MDM2 and VEGF inhibitor further contributes to its antitumor effects, making gossypol a potential therapeutic agent in combating age-related diseases and cancer.
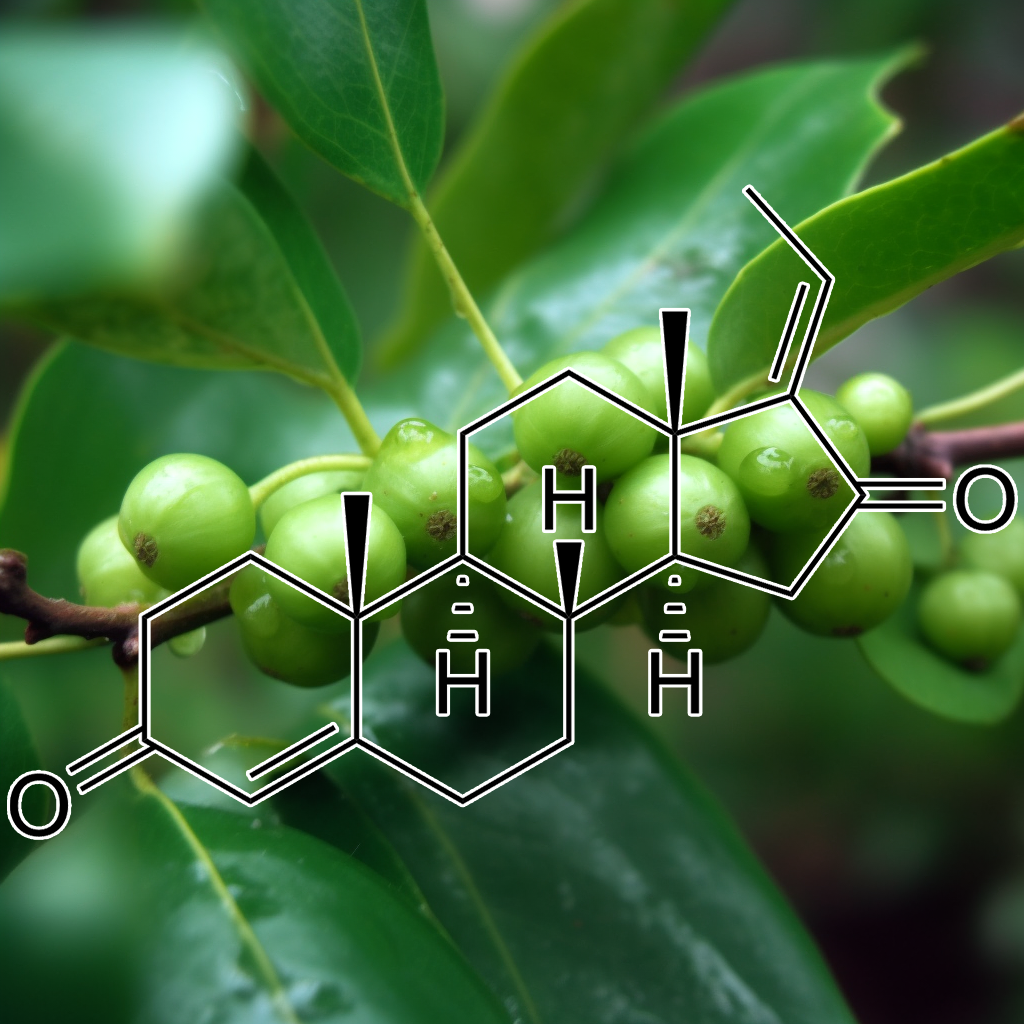
Guggulsterone

HESPERIDIN
Hesperidin, a bioflavonoid found in citrus fruits, shows potential as a senolytic agent targeting senescent cells. It modulates pathways like PI3K/AKT, mTOR, and Nrf2, promoting autophagy and apoptosis. Hesperidin may influence SASP (Senescence-Associated Secretory Phenotype), reduce inflammation, and regulate BCL-2 family proteins, enhancing cellular health. Its anti-inflammatory and antioxidant properties support longevity by mitigating cellular stress, improving metabolic functions, and promoting tissue regeneration, positioning hesperidin as a promising compound in healthy aging research.
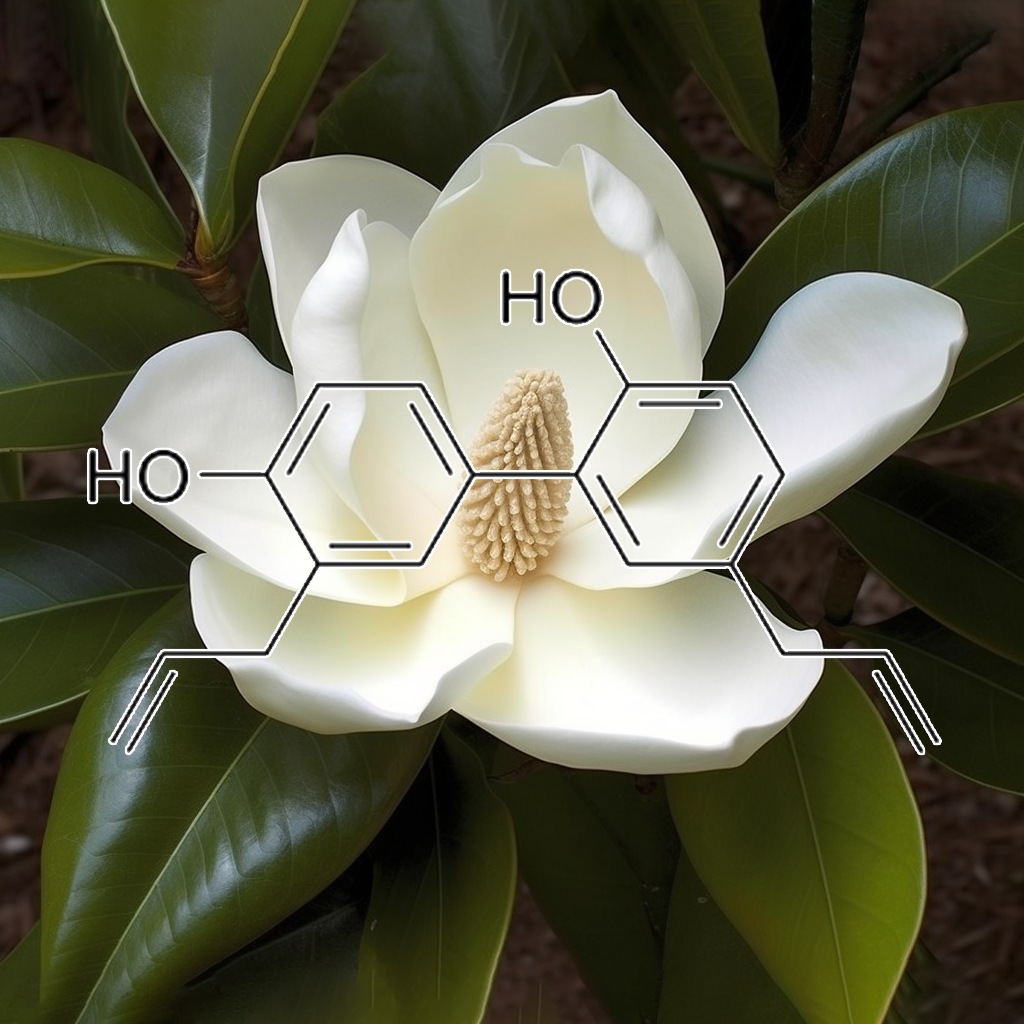
Honokiol, a bioactive compound from Magnolia bark, demonstrates significant health benefits by targeting senescent cells and modulating key apoptosis pathways. It inhibits STAT3 and NF-κB, disrupts tumor growth, and promotes autophagy via the PI3K/Akt/mTOR pathway. Honokiol also influences Bcl-2 family proteins, enhancing apoptosis through Bcl-XL downregulation. Additionally, it reduces the senescence-associated secretory phenotype (SASP), supporting cellular health and longevity. This compound offers promising potential in cancer prevention and therapy, highlighting its multifaceted therapeutic properties.


Jaceosidin is a flavonoid compound linked to anti-aging benefits by targeting senescent cells and reducing SASP (senescence-associated secretory phenotype). Its interaction with senolytic pathways, including PI3K/AKT, BCL-2, and mTOR, may promote apoptosis and autophagy, improving cellular health. Additionally, it may influence pathways like Nrf2 and cGAS-STING, reducing inflammation and oxidative stress. These effects, combined with its impact on apoptosis regulators (Bax, Bcl-xl) and heat shock proteins (HSP70, HSP90), support longevity and enhanced healthspan.
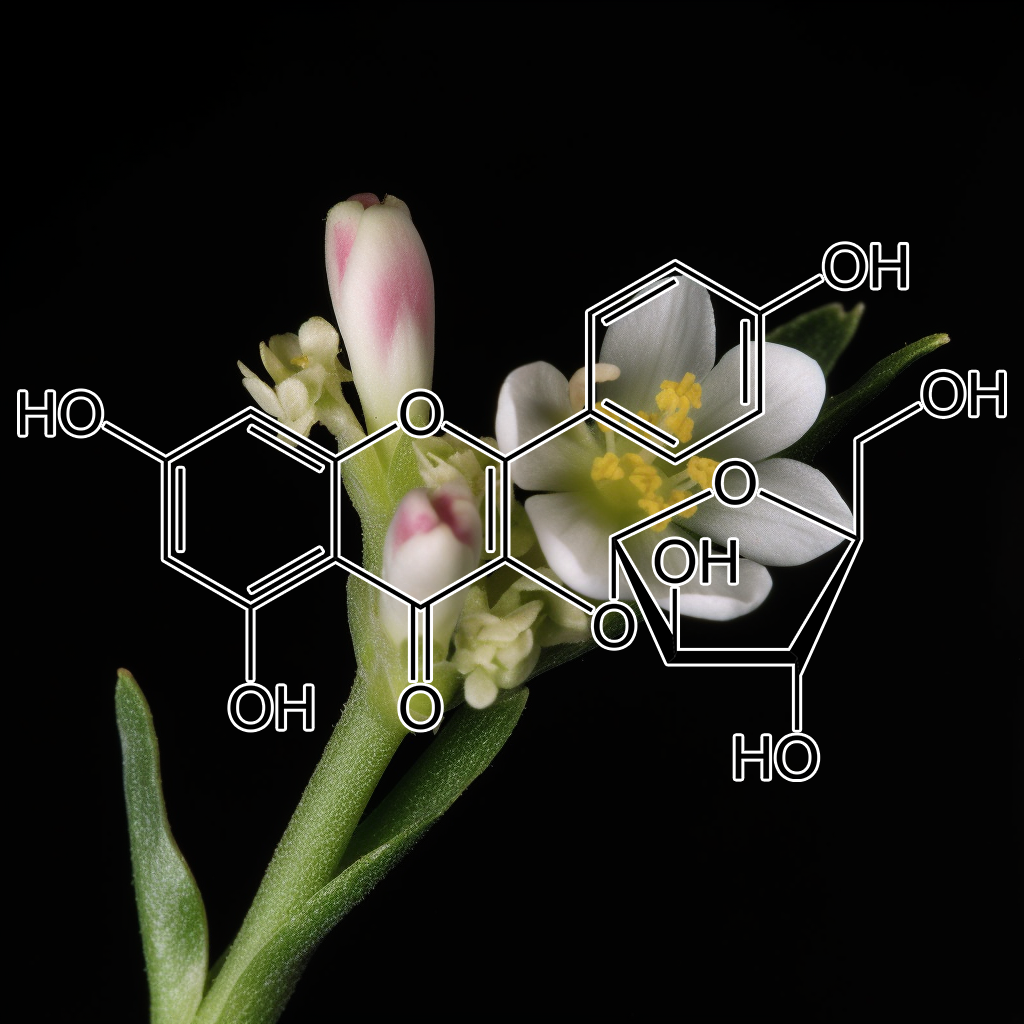
Juglanin
Juglanin, a natural flavonoid, has potential senolytic properties, influencing pathways like PI3K/AKT, Bcl-2, and mTOR. It targets senescent cells, potentially reducing SASP factors and promoting apoptosis via BAX, BAK, and caspases like CASPASE 3. Juglanin may modulate autophagy, reduce inflammation through Nrf2 and NF-κB, and interact with heat shock proteins like HSP70. These actions suggest Juglanin’s role in enhancing longevity by clearing senescent cells and promoting cellular health, contributing to anti-aging research.

Icariin exhibits promising senolytic properties by targeting senescent cells, influencing apoptosis pathways, and modulating critical proteins like Bcl-2 and caspases. It inhibits STAT3 activation, reducing anti-apoptotic factors, and promotes apoptosis in cancer cells while modulating autophagy and Nrf2 activation. This multifaceted approach may attenuate the senescence-associated secretory phenotype (SASP), contributing to cellular rejuvenation and improved health outcomes. Research supports its potential role in enhancing longevity and vitality through these mechanisms.
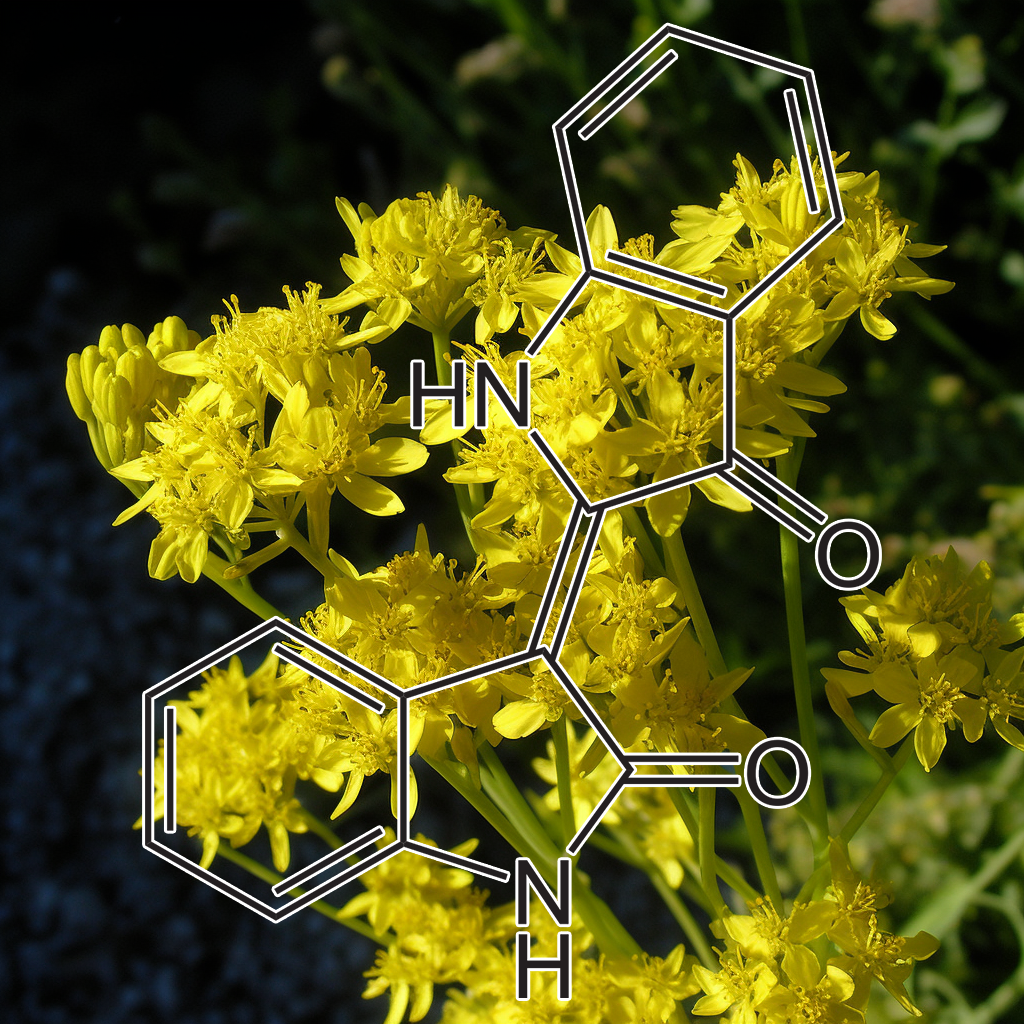
INDIRUBIN (INDIGO BLUE EXTRACT)
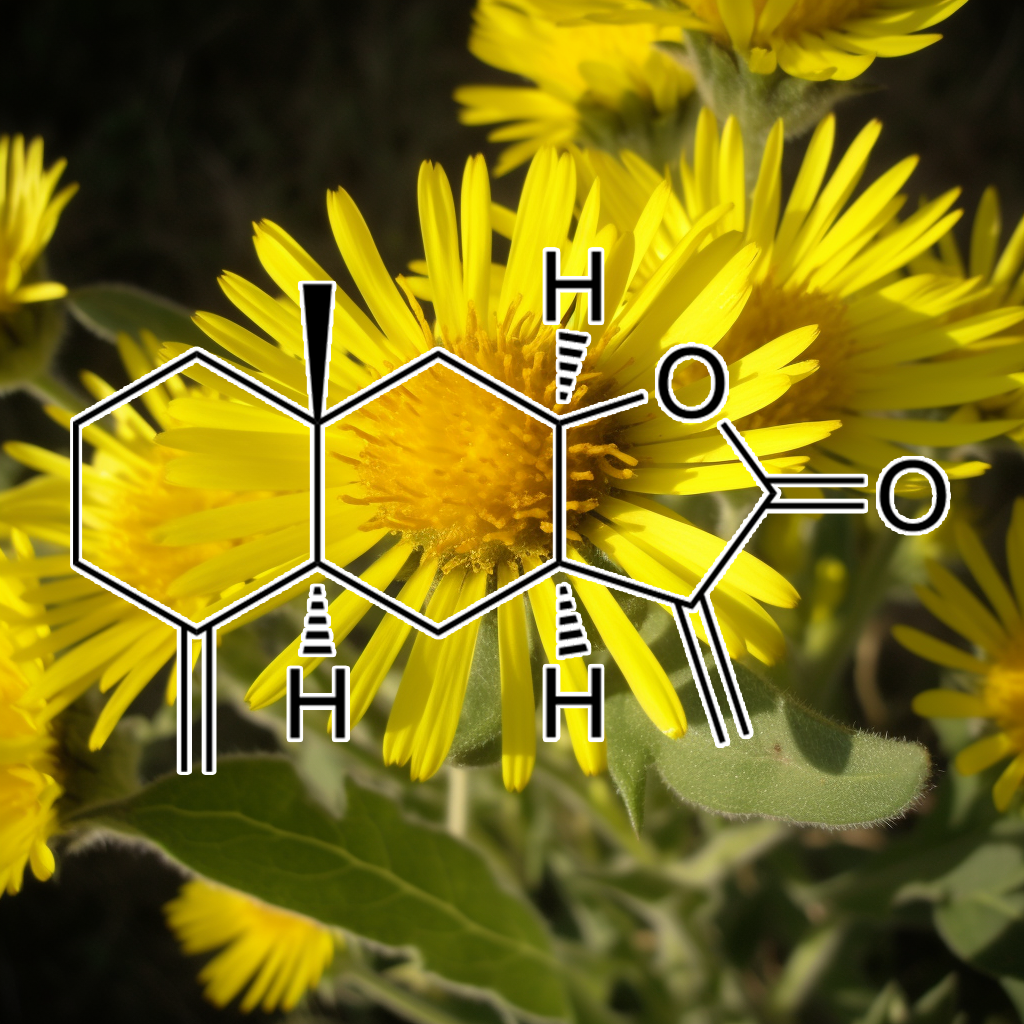
Isoalantolactone (Tumuxiang)
Isoalantolactone, a bioactive sesquiterpene lactone, exhibits potential senolytic properties by targeting senescent cells and associated pathways, including PI3K/AKT, mTOR, Bcl-2 family proteins (Bcl-xl, Bax, BAK), and apoptosis-related factors. It modulates cellular mechanisms like autophagy, STAT3, NF-kB, and cGAS-STING, which play crucial roles in cell senescence, inflammation (SASP), and aging. Emerging research suggests Isoalantolactone could support healthy aging by influencing apoptosis and cellular longevity pathways, though further studies are needed to confirm its senolytic efficacy.
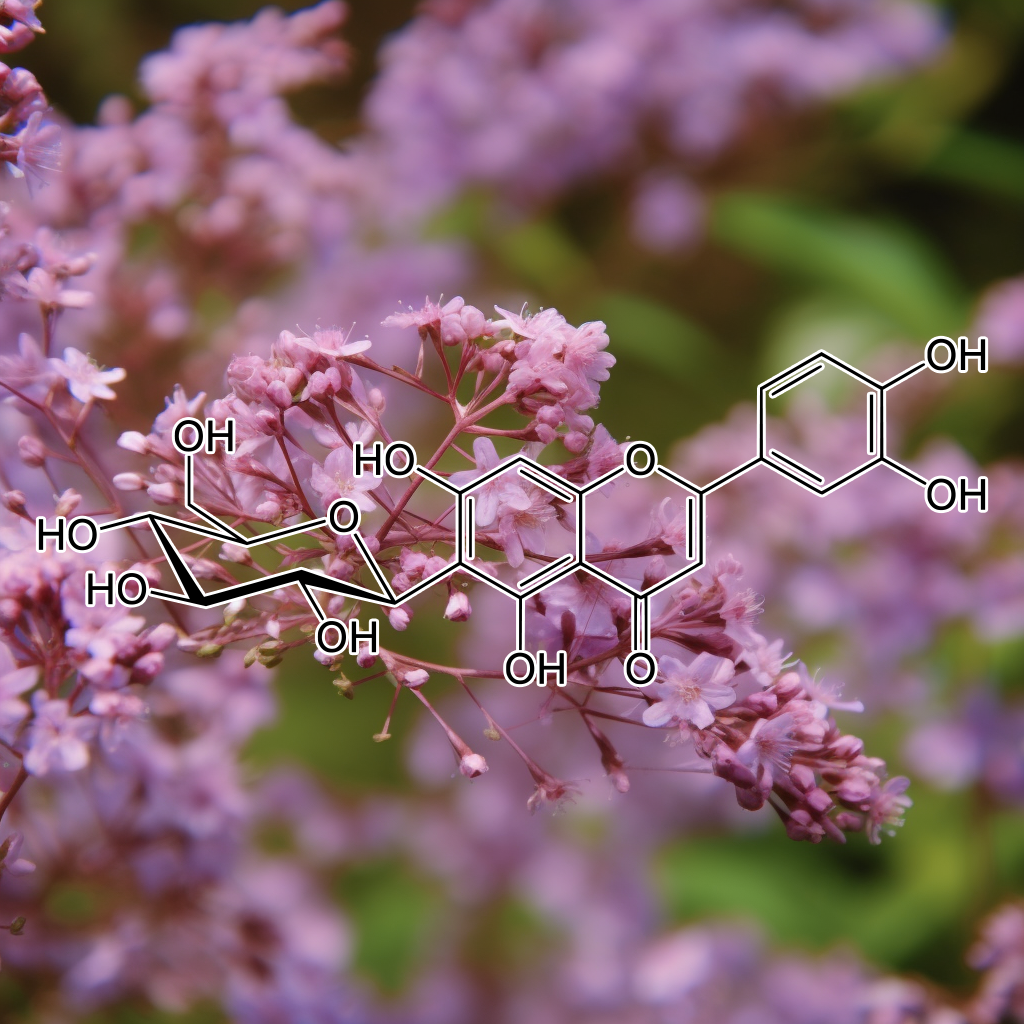
Targeting senescent cells and modulating pathways like PI3K/AKT, mTOR, and Nrf2, this plant-derived flavonoid has demonstrated potential senolytic effects. It may inhibit the SASP (senescence-associated secretory phenotype) and promote apoptosis through Bcl-2 family proteins. Additionally, it influences autophagy, NF-kB, and STAT3, helping to reduce inflammation, enhance longevity, and support cellular health. Its role in clearing senescent cells positions it as a promising agent for anti-aging and healthspan-extending therapies.
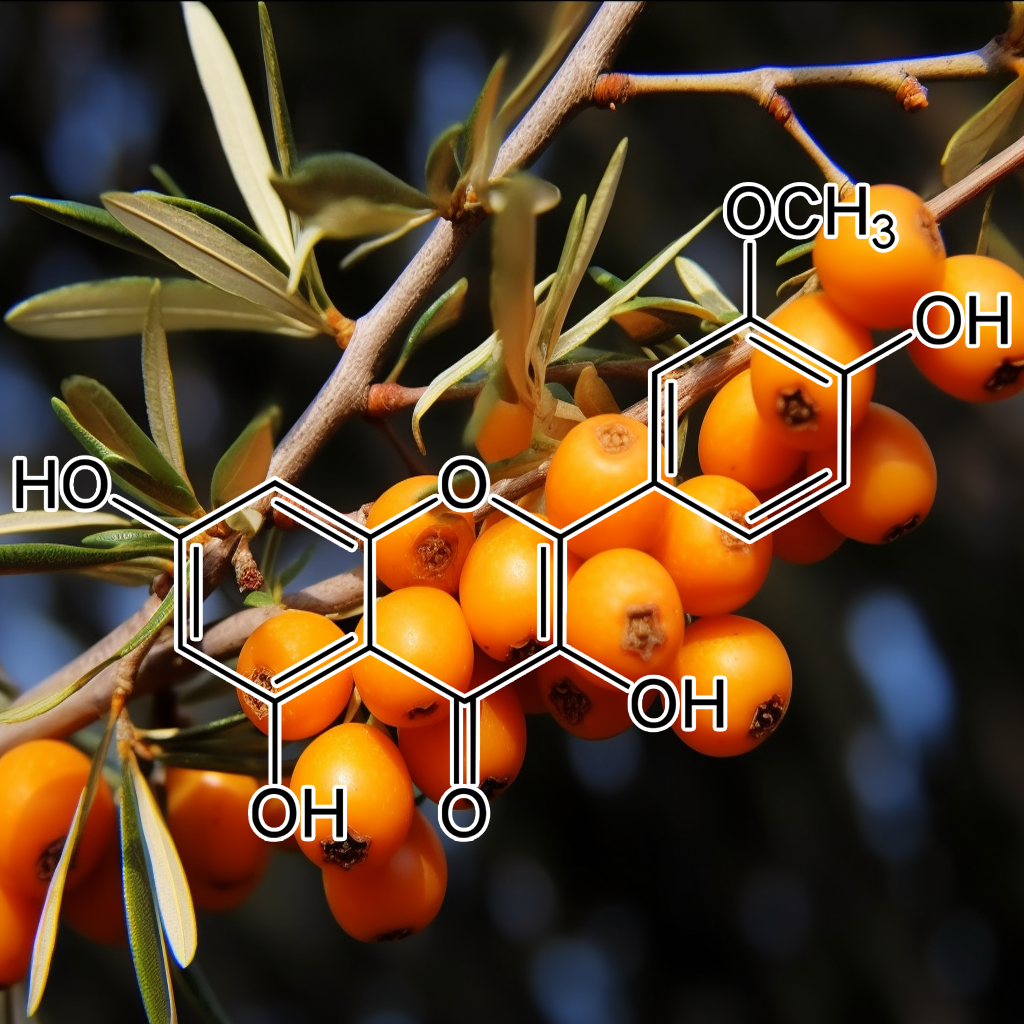
Isorhamnetin is a flavonoid with proven senolytic properties, targeting senescent cells and modulating pathways like PI3K/AKT, BCL-2, and mTOR. It reduces SASP, enhances autophagy, and supports apoptosis regulation via BAX and BCL-xL. Its influence on STAT3, Nrf2, and NF-kB contributes to reduced inflammation and oxidative stress. This compound has demonstrated potential to enhance healthspan by targeting senescence-associated pathways, promoting cellular repair, longevity, and overall health optimization.

This natural flavonoid shows potential senolytic effects by targeting senescent cells and influencing key aging-related pathways, such as PI3K/AKT, BCL-2, Nrf2, mTOR, and autophagy. It helps regulate apoptosis, reduce SASP (senescence-associated secretory phenotype), and enhance cellular health and longevity. Evidence supports its role in modulating pathways like STAT3, NFKB, and caspase 3, boosting cellular resilience against oxidative stress and inflammation, potentially contributing to anti-aging and longevity therapies.

Kaempferol is a natural flavonoid with senolytic properties, targeting senescent cells and mitigating their harmful effects, such as SASP secretion. It influences pathways like PI3K/AKT, BCL-2, and mTOR, promoting apoptosis and autophagy. Kaempferol also modulates Nrf2, TLR4, and STAT3, reducing inflammation and oxidative stress. Its ability to impact proteins like BAX, BCL-XL, and caspase-3 highlights its role in cellular homeostasis, supporting longevity and healthy aging by clearing damaged cells and enhancing cellular function.

Kurarinone, a natural flavonoid, is linked to cellular senescence by targeting senolytic pathways such as PI3K/AKT and BCL-2 family proteins. It modulates apoptosis and autophagy, potentially reducing senescence-associated secretory phenotype (SASP). Its impact on pathways like mTOR and Nrf2 supports cellular health and longevity by promoting the clearance of senescent cells. Kurarinone’s anti-inflammatory and antioxidant properties make it a promising compound for healthy aging, targeting mechanisms like STAT3, NF-κB, and caspase-3 activation.

Targeting senescent cells through key pathways like PI3K/AKT, BCL-2, and cGAS-STING can promote longevity and healthspan. This compound modulates autophagy, mTOR, and Nrf2 pathways, influencing cellular senescence and apoptosis. It also impacts the balance of pro- and anti-apoptotic proteins, including BAX and BCL-xl, while regulating caspase activation and NF-κB signaling. These effects are linked to reducing age-related diseases, inflammation, and improving overall cellular health.

Lipoic acid is a potent antioxidant known to modulate cellular pathways related to senescence and longevity. It influences senolytic activity by targeting senescent cells and modulating key pathways like PI3K/AKT, mTOR, and Nrf2. Lipoic acid also interacts with apoptosis regulators (Bcl-2 family), autophagy processes, and inflammatory signals like NF-kB, cGAS-STING, and STAT3. These effects promote cellular health, reduce SASP (senescence-associated secretory phenotype), and enhance longevity by maintaining mitochondrial function and reducing oxidative stress.
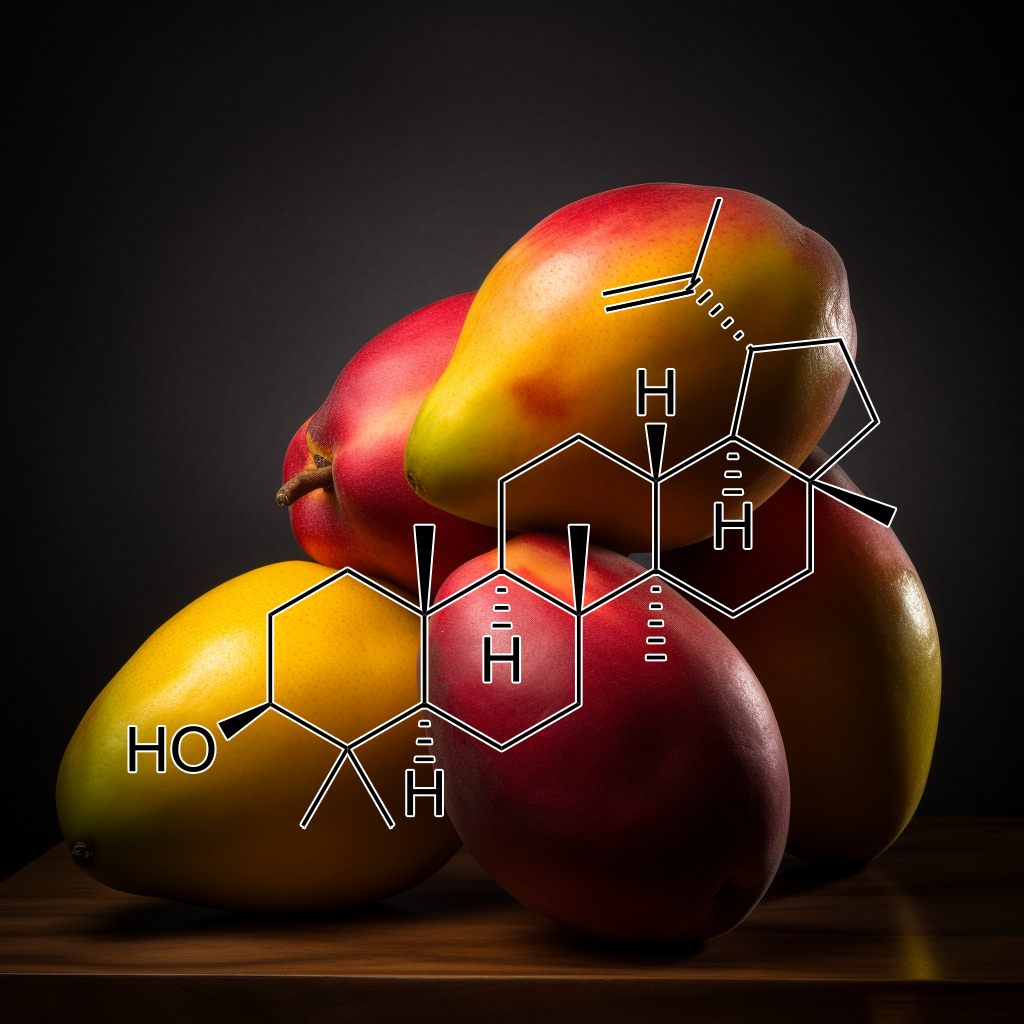
Lupeol has shown potential in targeting senescent cells and influencing senolytic pathways such as PI3K/AKT, BCL-2, mTOR, and autophagy. It may reduce SASP (senescence-associated secretory phenotype) factors, enhance apoptosis, and modulate inflammation through pathways like NF-κB, STAT3, and TLR4. Lupeol’s interactions with proteins such as Bcl-2, Bax, and caspases contribute to cellular longevity and improved health outcomes. Its role in targeting aging mechanisms makes it promising for enhancing lifespan and cellular resilience.
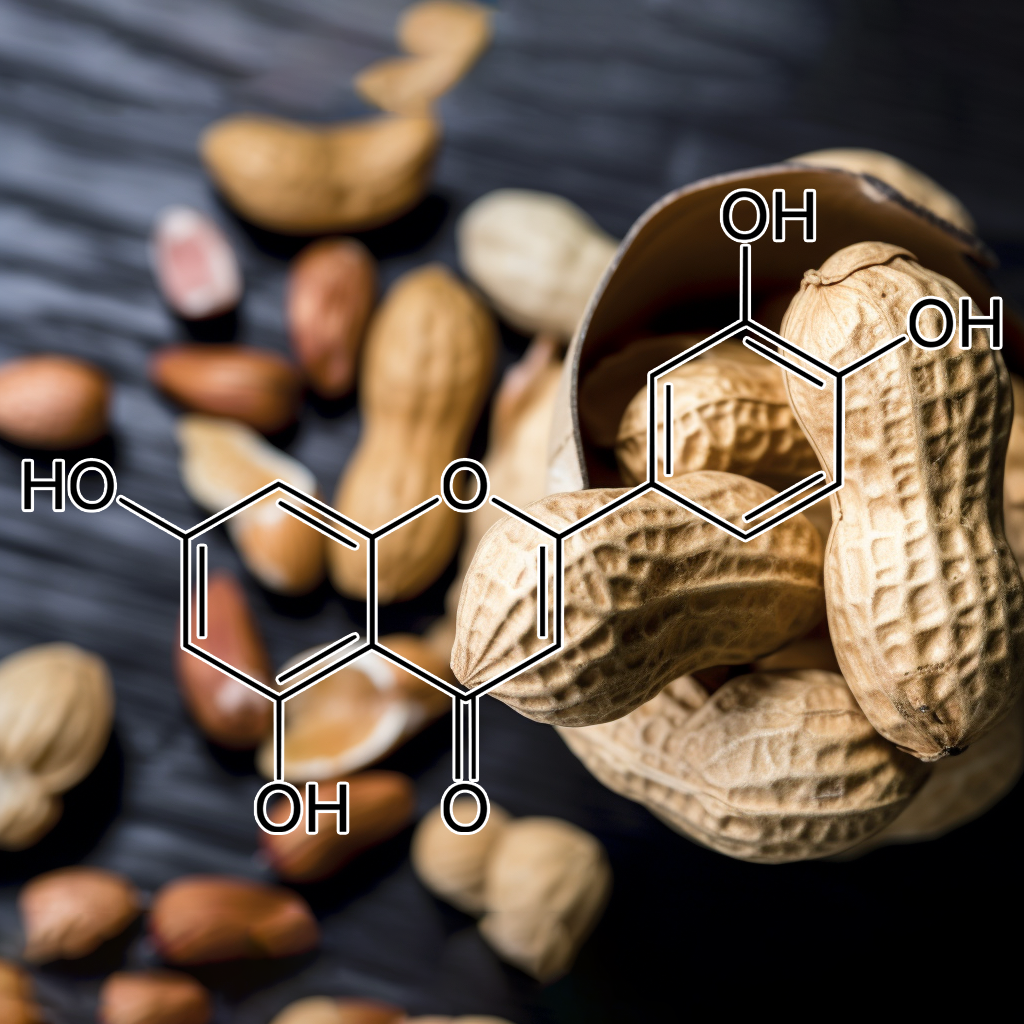
A natural flavonoid exhibits senolytic activity by targeting senescent cells and reducing SASP (senescence-associated secretory phenotype). It modulates key pathways like PI3K/AKT, mTOR, Nrf2, and BCL-2 family proteins, influencing autophagy, apoptosis, and cellular stress responses. Linked to reduced inflammation and enhanced cellular health, it promotes longevity by aiding in senescent cell clearance. Potential effects on STAT3, NF-κB, and caspase-3 further highlight its role in healthy aging and disease prevention.

LYCOPENE
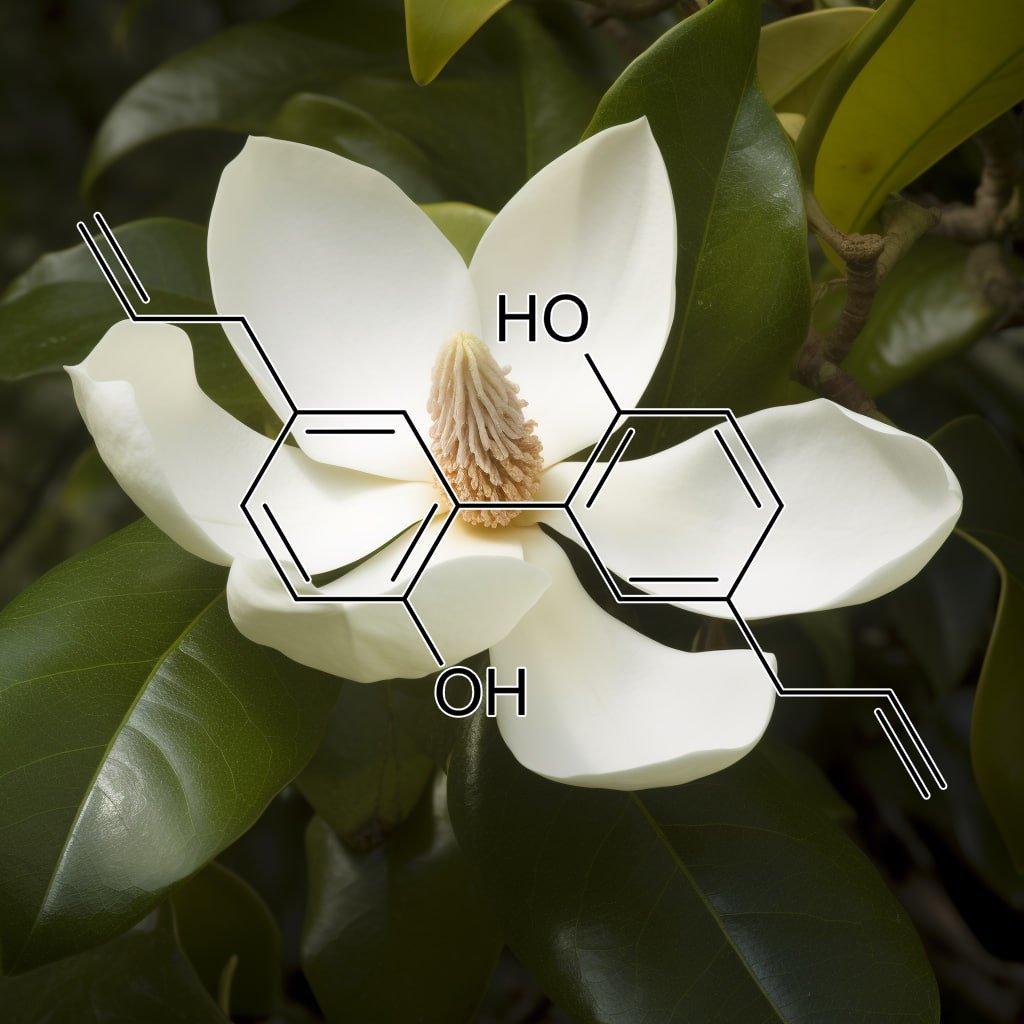

Mulberry (Morus alba) contains bioactive compounds that may have senolytic properties, targeting senescent cells to improve health and longevity. Research shows potential in influencing pathways like PI3K/AKT, mTOR, Nrf2, and autophagy. It also affects apoptosis regulators like Bcl-2, Bax, and caspases. These actions reduce harmful SASP factors, aiding in cellular rejuvenation and promoting healthy aging. Morus alba may also impact oxidative stress response and inflammation, supporting overall wellness and longevity through cellular health optimization.
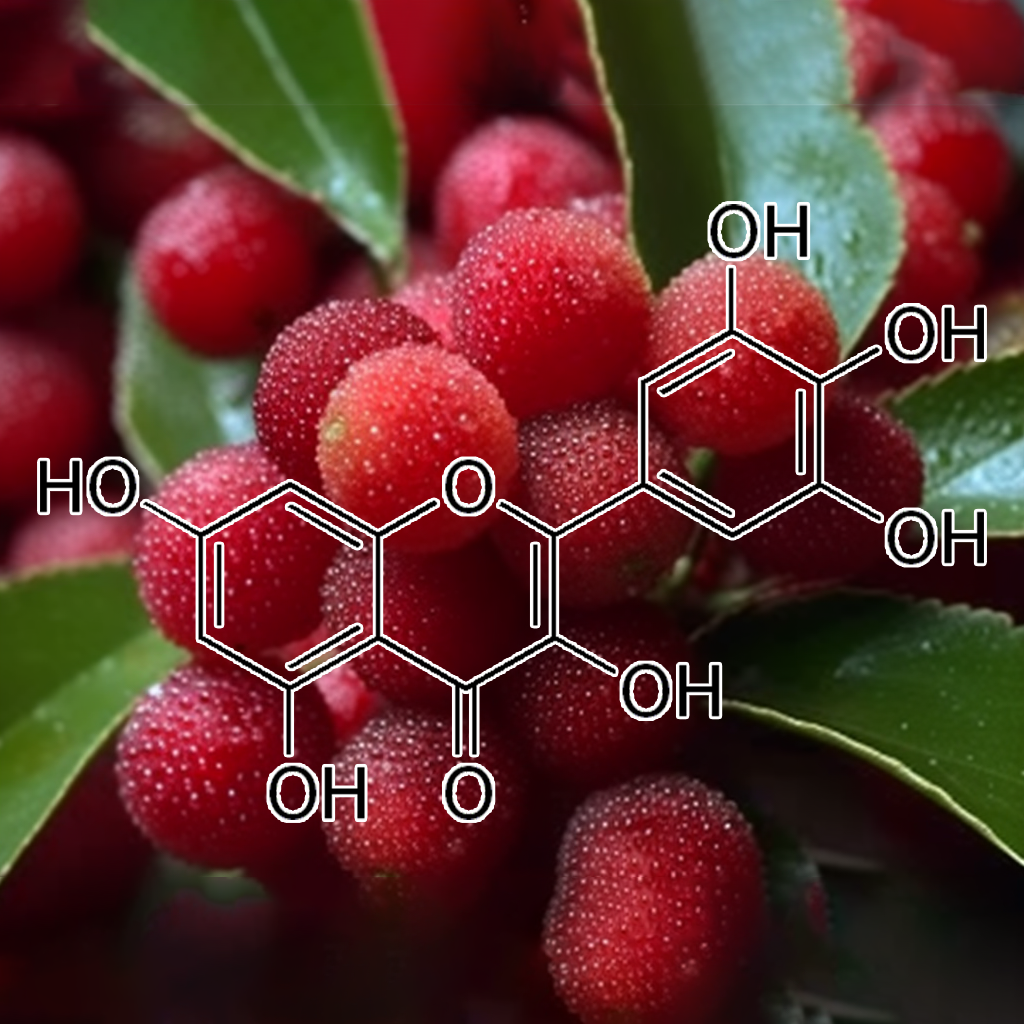
Myricetin, a natural flavonoid, has shown promising effects in targeting senescent cells and modulating senolytic pathways, including PI3K/AKT, BCL-2, mTOR, and autophagy. It aids in reducing SASP (senescence-associated secretory phenotype) and promotes apoptosis through pathways like BAX, PUMA, and CASPASE 3. Additionally, it influences cellular stress responses via Nrf2 and cGAS-STING. By mitigating senescence, Myricetin supports longevity and healthspan, offering potential benefits for age-related diseases and cellular repair.
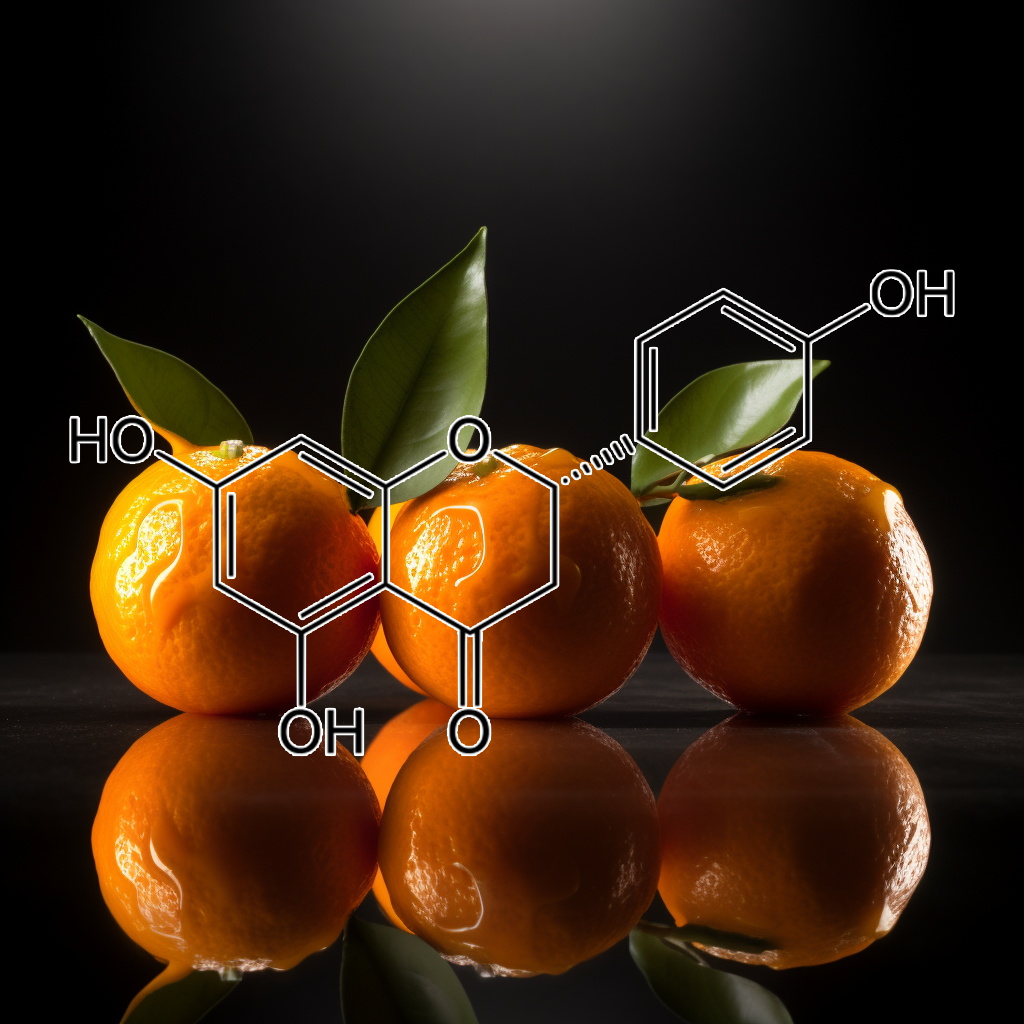
Naringenin has demonstrated senolytic properties by targeting senescent cells. It modulates key pathways like PI3K/AKT, Nrf2, and mTOR, contributing to reduced inflammation and improved cellular health. Naringenin also affects SASP, apoptosis regulators (Bcl-2, Bax), and autophagy processes. Evidence suggests it promotes longevity by enhancing cellular repair and preventing chronic disease through mechanisms like cGAS-STING, STAT3, and NFKB, with potential benefits in aging-related conditions.

Neem Leaf (Azadirachta indica)
Neem leaf has been studied for its potential senolytic effects, targeting senescent cells and promoting healthy aging. It influences pathways like PI3K/AKT, BCL-2, and mTOR, and is linked to autophagy, apoptosis, and the regulation of proteins such as BAX, BCL-XL, and caspases. Neem leaf may modulate inflammatory responses, Nrf2 activation, and senescence-associated secretory phenotype (SASP), supporting cellular health and longevity. Its interaction with key survival pathways offers promise for anti-aging and regenerative therapies.
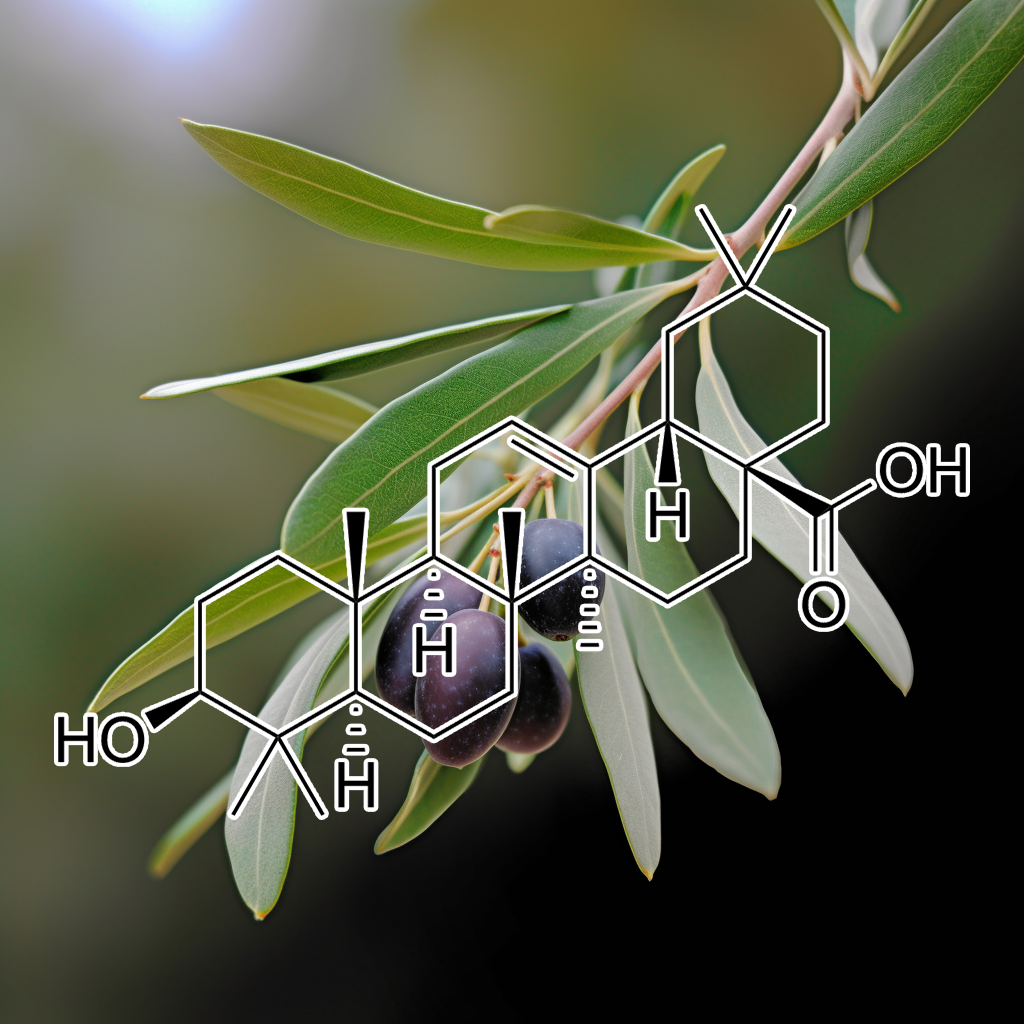
Oleacein and Oleanolic acid
Oleacein and oleanolic acid are natural compounds with potential senolytic properties, targeting senescent cells and pathways like PI3K/AKT, BCL-2, mTOR, and autophagy. These compounds may modulate SASP, apoptosis regulators (BAX, BCL-xL, PUMA), and cellular stress responses like Nrf2 and cGAS-STING. By influencing key pathways and proteins (e.g., CASPASE 3, STAT3, NF-κB, HSPs), they support cellular health and longevity, offering potential therapeutic benefits for aging and age-related diseases.

Oleuropein
Oleuropein, a compound in olive leaves, has shown potential senolytic effects by targeting senescent cells and influencing pathways like PI3K/AKT, BCL-2, and mTOR. It may reduce inflammation, modulate autophagy, and enhance apoptosis through interactions with proteins such as BAX, BAK, and BIM. Oleuropein also impacts pathways like NF-κB, STAT3, and Nrf2, potentially promoting longevity and healthy aging by clearing senescent cells and reducing the harmful effects of the SASP (senescence-associated secretory phenotype).
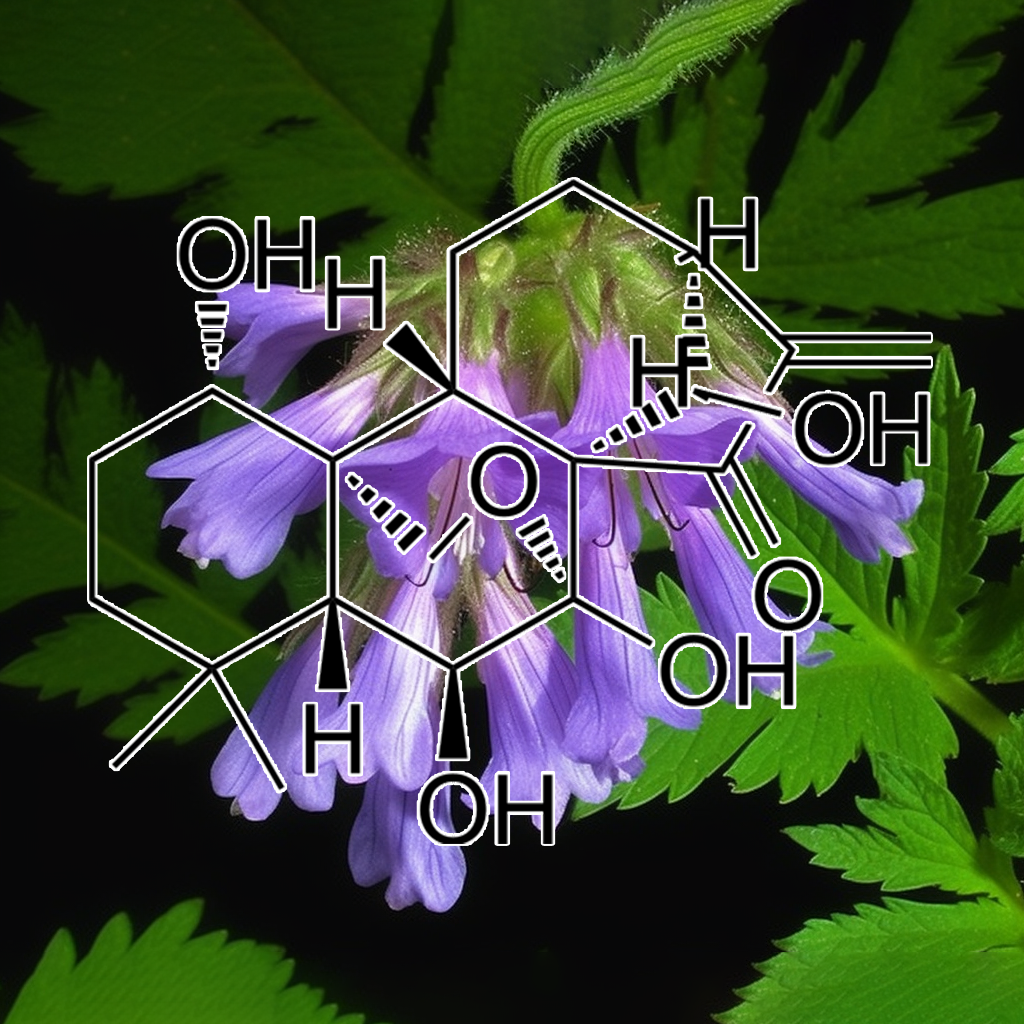
Oridonin, a bioactive compound from Rabdosia rubescens, exhibits senolytic properties by targeting senescent cells and associated pathways, such as PI3K/AKT, BCL-2, and mTOR. It promotes apoptosis and autophagy, modulates SASP, and inhibits inflammatory pathways like NF-kB and STAT3. Oridonin also interacts with key proteins like BAX, BAK, PUMA, and caspases, enhancing cell death in senescent cells. Its potential to improve healthspan and longevity by targeting cellular senescence is supported by emerging scientific evidence.
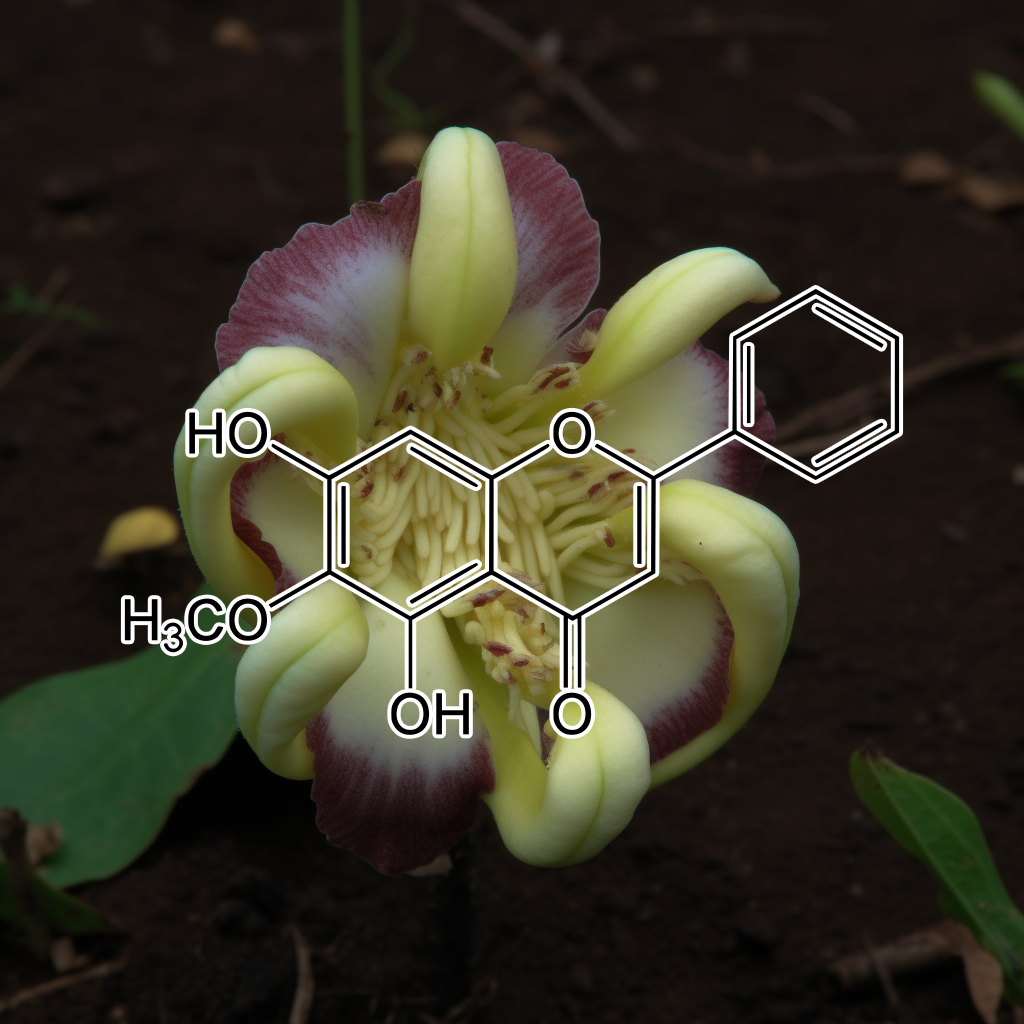
Oroxylin A
Oroxylin A is a bioactive flavonoid with emerging senolytic potential, promoting health and longevity by targeting senescent cells. It influences key pathways such as PI3K/AKT, mTOR, Nrf2, and apoptosis regulators like Bcl-2 and Bcl-xl. Oroxylin A modulates SASP (senescence-associated secretory phenotype), reduces inflammation via STAT3 and NF-kB, and enhances autophagy. These effects help combat age-related dysfunction, cellular senescence, and support overall healthy aging. It also impacts apoptosis via caspases, promoting cell renewal and longevity.
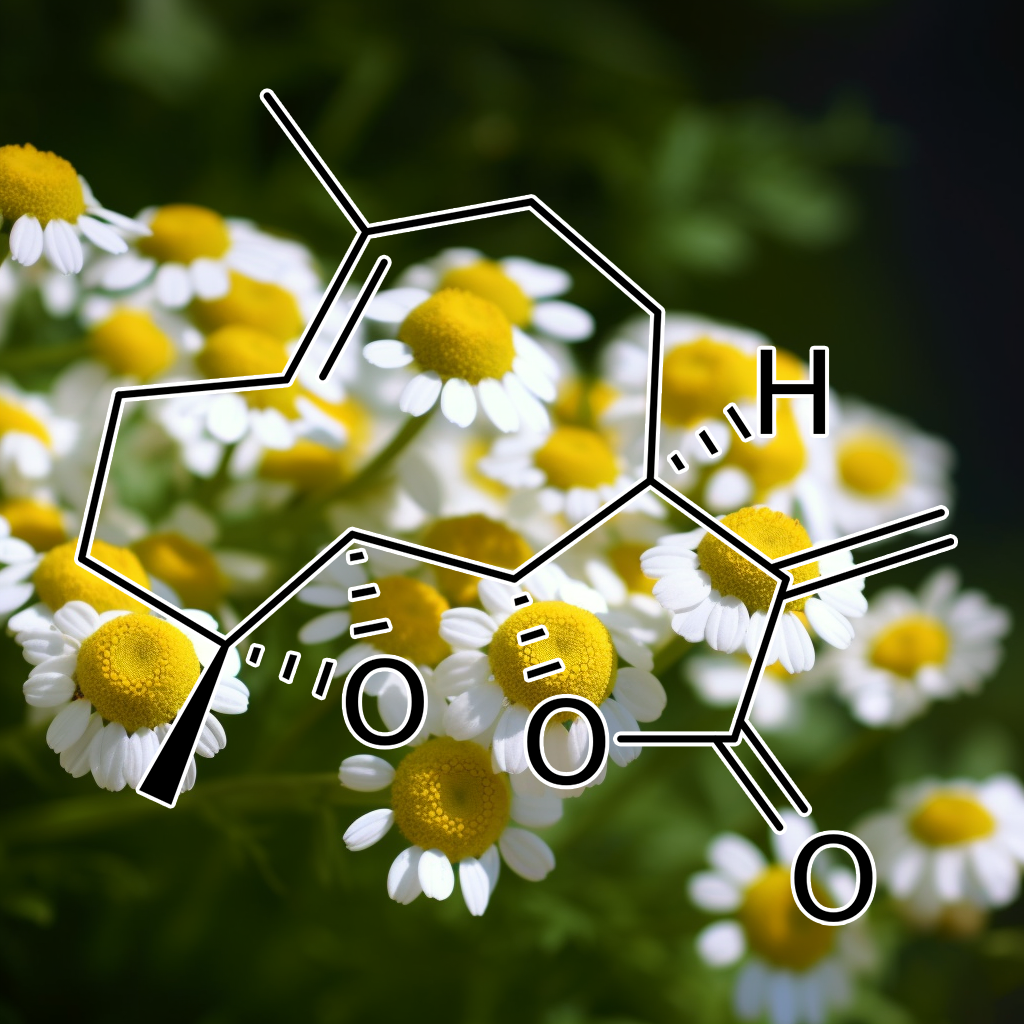

Patrinia scabiosaefolia
Patrinia scabiosaefolia has demonstrated potential in targeting senescent cells through senolytic pathways like PI3K/AKT, BCL-2, mTOR, and autophagy. It may impact apoptosis-related proteins (e.g., BAX, BCL-xl) and inflammation via STAT3 and NF-κB. Research highlights its role in promoting cell death in damaged cells, potentially reducing SASP (Senescence-Associated Secretory Phenotype) and enhancing longevity. These effects are linked to improved healthspan and cellular rejuvenation, supporting the body’s ability to counter aging-related decline.
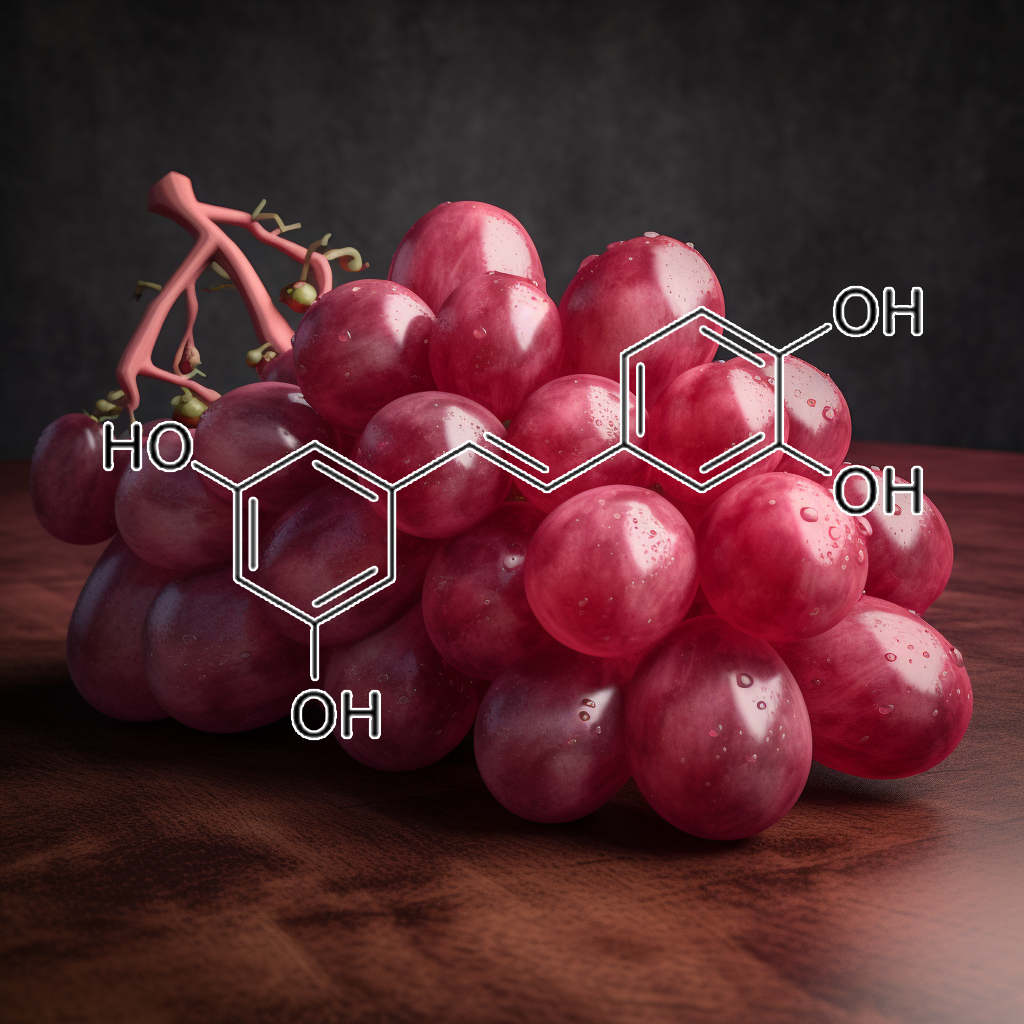
Piceatannol, a natural polyphenol, shows potential as a senolytic, targeting senescent cells by modulating pathways like PI3K/AKT, mTOR, and BCL-2 family proteins. It inhibits SASP factors, enhancing autophagy, apoptosis, and reducing inflammation via pathways such as NF-κB and STAT3. Piceatannol’s role in regulating senescence-associated pathways like cGAS-STING and Nrf2 suggests its promise for promoting healthy aging and longevity, aligning with health optimization trends in cellular senescence research.

Phloretin, an antioxidant derived from apples, demonstrates significant senolytic potential by targeting and eliminating senescent cells, which contribute to aging and age-related diseases. It induces apoptosis through the JNK1/2 and p38 MAPK pathways, regulating key proteins like Bcl-2 and promoting the activation of cleaved caspases. Additionally, phloretin inhibits glucose transporters, addressing metabolic demands in senescent cells. These mechanisms suggest phloretin’s promise in enhancing health, supporting longevity, and combating age-related conditions.

Phloroglucinol
Phloroglucinol shows promise in targeting senescent cells, linked to aging and disease. It potentially modulates key pathways like PI3K/AKT, mTOR, and Nrf2, influencing apoptosis, autophagy, and SASP reduction. By interacting with proteins like Bcl-2, Bax, and caspases, it may induce cell death in senescent cells. This action could enhance health and longevity by improving tissue function and reducing inflammation. Research on its senolytic effects is ongoing, but its role in cellular health is increasingly recognized.

Phosphatidylcholine
Phosphatidylcholine, a key component of cell membranes, supports cellular health by promoting repair and regeneration. It may act as a senolytic by helping to eliminate senescent (aging) cells, potentially improving tissue function, reducing inflammation, and promoting healthy aging. Phosphatidylcholine (PPC) triggers apoptosis in 3T3-L1 adipocytes by activating stress-related signaling proteins, unlike sodium deoxycholate, which causes cell lysis and potential necrosis.

Phyllanthus emblica
Phyllanthus emblica (Indian gooseberry) is linked to cellular senescence pathways like PI3K/AKT, BCL-2, and Nrf2. It may promote autophagy, reduce SASP factors, and influence apoptosis-related proteins (BAX, Bcl-xl) to clear senescent cells, supporting longevity, reducing inflammation, and enhancing cellular health through senolytic actions. Phyllanthus emblica extract induces apoptosis, reduces fat accumulation, and inhibits adipogenesis via molecular pathways, offering potential therapeutic benefits for obesity treatment.
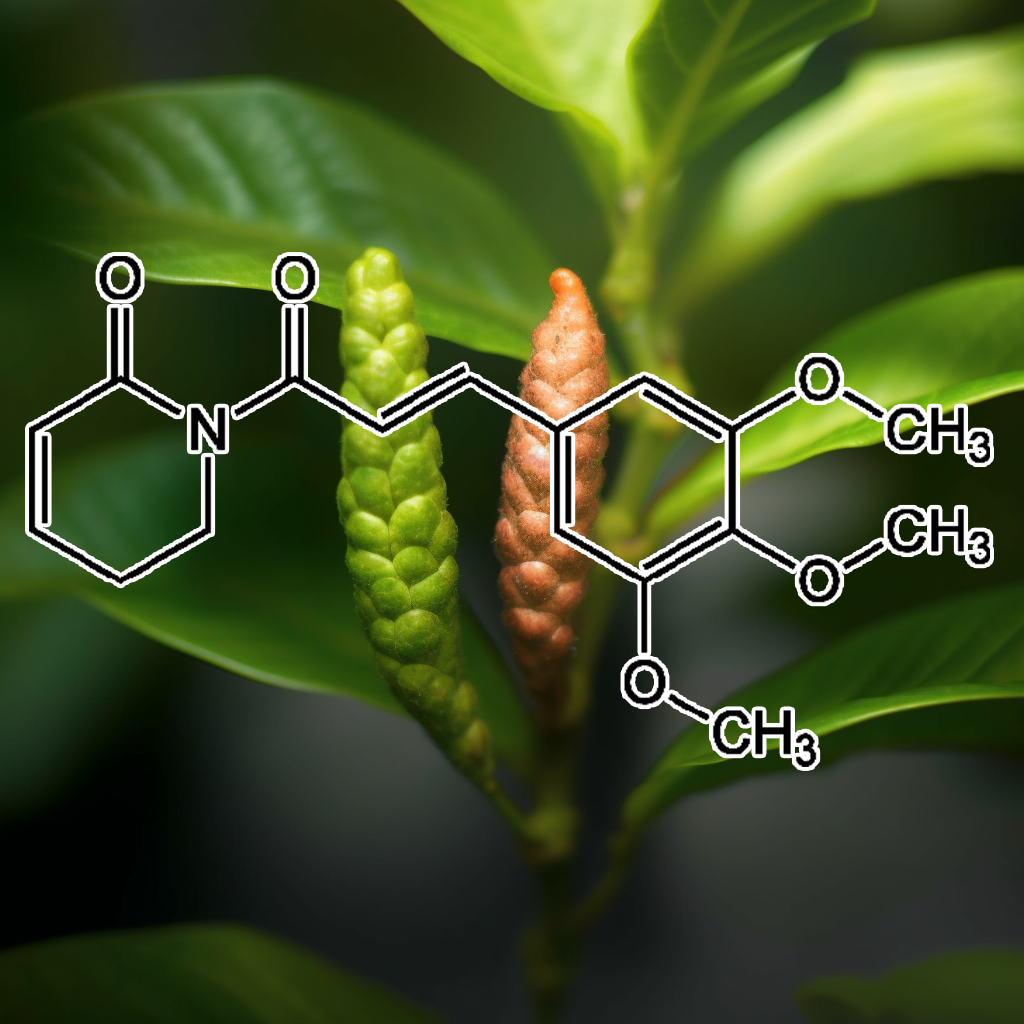
Piperlongumine exhibits senolytic properties, selectively inducing apoptosis in senescent cells by modulating critical pathways such as PI3K/AKT, mTOR, and Nrf2. This compound reduces the senescence-associated secretory phenotype (SASP) and enhances autophagy, promoting longevity. Its role in activating the JNK pathway and targeting STAT3-positive cancer cells further supports its potential in cancer therapy. By influencing apoptosis regulators like Bcl-2 and Bax, Piperlongumine offers promising health benefits, particularly in age-related diseases and inflammation.
Phloroglucinol exhibits potential senolytic activity by targeting senescent cells, modulating pathways like PI3K/AKT, mTOR, Nrf2, and Bcl-2. It may inhibit SASP factors, reduce inflammation via NF-κB and STAT3, and promote apoptosis through caspases. These effects support cellular health, anti-aging, and longevity by minimizing senescence-associated damage. Phloroglucinol induces apoptosis by modulating Bcl-2 family proteins, promoting mitochondrial dysfunction, and triggering apoptotic pathways in carcinoma cells through cytochrome c and Apaf-1 release.

POMEGRANATE PEEL
Pomegranate peel, rich in bioactive compounds, exhibits senolytic properties that target senescent cells, influencing key longevity pathways such as PI3K/AKT, mTOR, and NF-kB. These compounds reduce the senescence-associated secretory phenotype (SASP) and modulate inflammation by inhibiting TLR4 and pro-inflammatory mediators. Ellagic acid further enhances skin health by blocking collagen degradation and promoting antioxidant pathways. Collectively, these effects support cellular health, autophagy, and apoptosis regulation, potentially contributing to enhanced longevity and overall well-being.
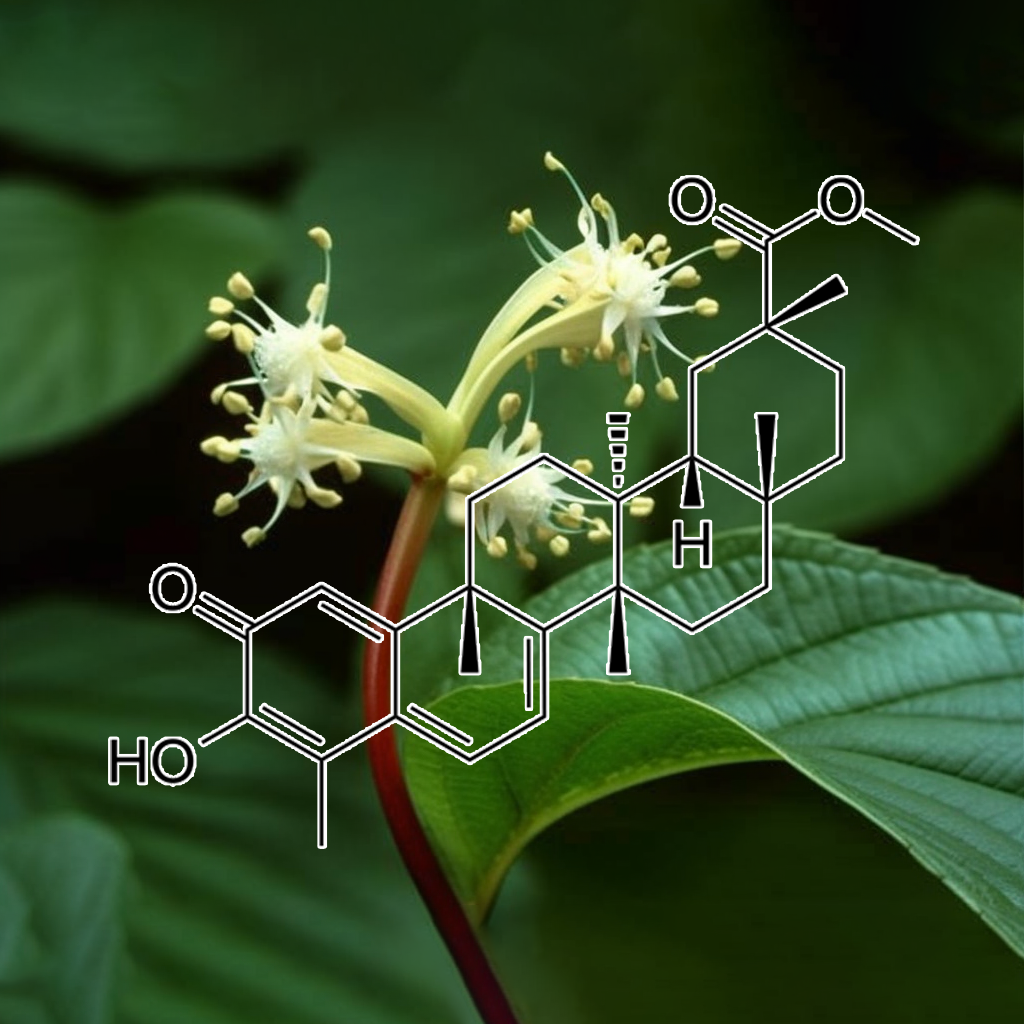
Pristimerin, a triterpenoid, targets senescent cells through pathways like PI3K/AKT, BCL-2, and mTOR, promoting apoptosis and reducing the senescence-associated secretory phenotype (SASP). It effectively inhibits pro-survival proteins such as Bcl-2 and Bcl-xL, activates caspases, and enhances autophagy. Additionally, Pristimerin modulates inflammation by regulating NF-κB and STAT3, showcasing potential for anticancer effects, particularly in colon cancer, while supporting healthy aging and longevity through its multifaceted mechanisms.

Proanthocyanidins (GRAPE SEED)
Proanthocyanidins from grape seed show senolytic potential by targeting senescent cells and modulating key pathways like PI3K/AKT, Nrf2, and mTOR. They influence apoptosis (Bcl-2, BAX), autophagy, and inflammation (TLR4, NF-kB), promoting healthy aging. These compounds may also regulate heat shock proteins (HSPs), enhancing cellular repair and longevity. Grape seed proanthocyanidins (GSPs) induce apoptosis, promote G1 arrest, activate caspases, and inhibit tumor growth in non-small cell lung cancer models.
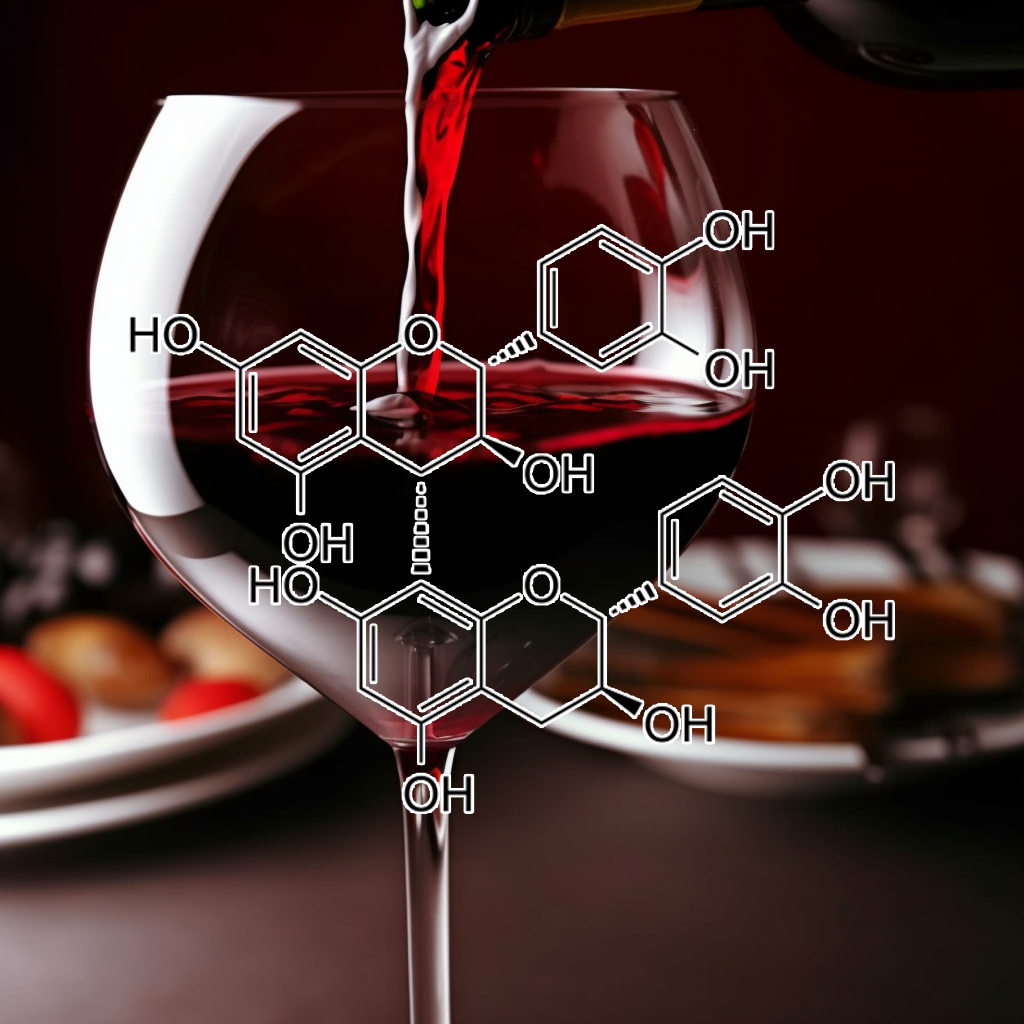
Procyanidin B3 targets senescent cells by modulating senolytic pathways like PI3K/AKT, BCL-2, and mTOR, promoting apoptosis and autophagy. It influences SASP, reduces inflammation via NF-kB, STAT3, and TLR4, and interacts with HSP proteins, caspases, and IAPs, enhancing health and longevity through cellular rejuvenation. Pro-B3 specifically targets and inhibits p300-mediated AR acetylation, limiting prostate cancer growth. It enhances flutamide’s effectiveness by disrupting acetylation-dependent cell proliferation and inducing cell death.

Pseudolaric acid B exhibits potential senolytic effects by targeting senescent cells, impacting pathways like PI3K/AKT, BCL-2, and autophagy. It modulates apoptosis via Bax, Bcl-xl, and caspases, reducing SASP and inflammation through cGAS-STING, Nrf2, STAT3, and NF-κB, promoting health and longevity. Pseudolaric acid B (PLAB) inhibits glioma cell growth by inducing apoptosis via p53 upregulation, BAX increase, BCL-2 suppression, and caspase activation, with minimal organ toxicity.

Pterostilbene, a natural compound found in blueberries, shows potential as a senolytic agent, targeting senescent cells to promote healthy aging. It modulates key pathways like PI3K/AKT, mTOR, and Nrf2, while influencing apoptosis-related proteins such as BCL-2, Bax, and PUMA. By reducing SASP and enhancing autophagy, pterostilbene may mitigate inflammation and cellular damage, potentially contributing to improved longevity and overall health. Further research is needed to fully establish its therapeutic potential.
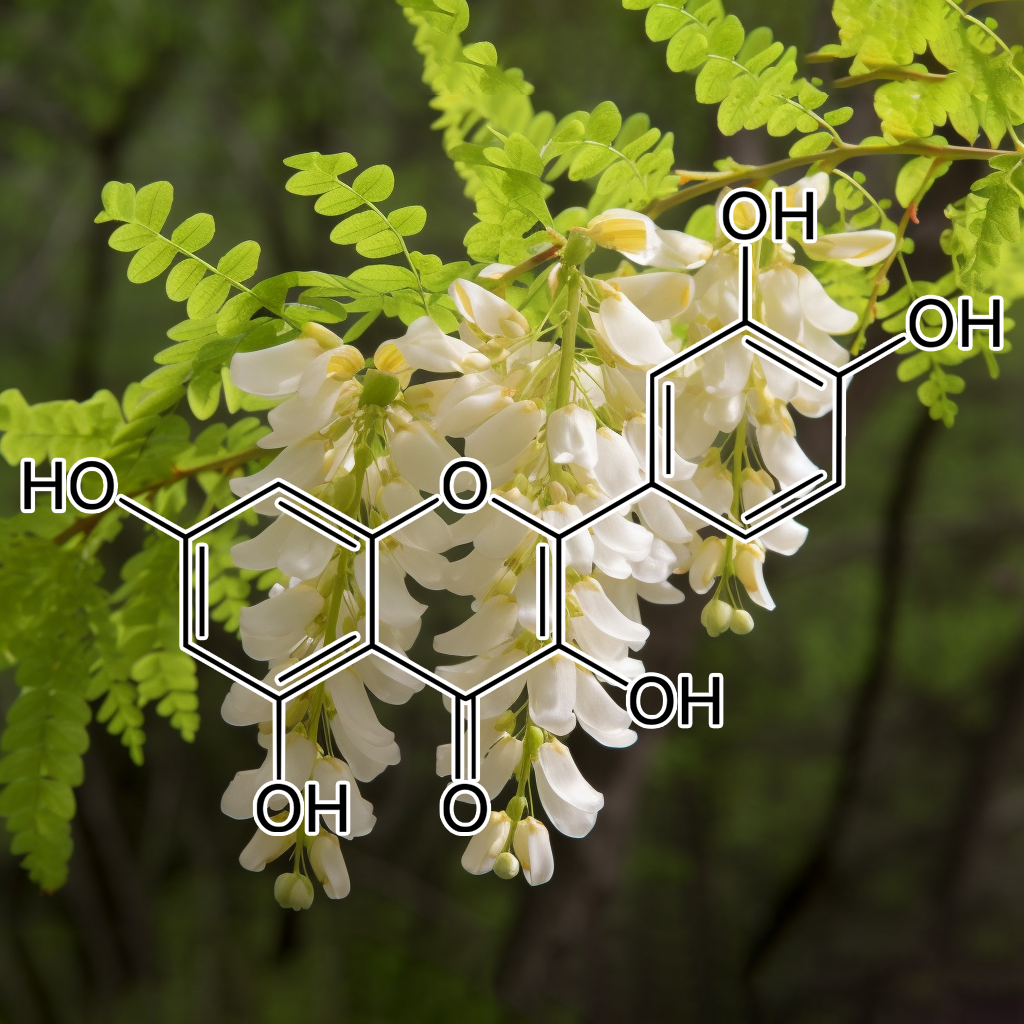
Quercetin
Quercetin, a potent flavonoid, exhibits senolytic properties by targeting senescent cells and modulating key pathways such as PI3K/AKT, BCL-2, and Nrf2. It promotes apoptosis in cancer cells while reducing the senescence-associated secretory phenotype (SASP), thereby inhibiting inflammatory markers like NF-kB and STAT3. This dual action not only combats aging and cancer but also enhances healthspan through improved autophagy and mTOR regulation. Quercetin’s therapeutic potential positions it as a valuable compound for anti-aging and cancer treatments.

Resveratrol has emerged as a promising compound in health science, targeting senescent cells and enhancing longevity. It modulates key pathways, including PI3K/AKT, mTOR, and BCL-2, promoting autophagy and apoptosis while reducing the senescence-associated secretory phenotype (SASP). By inhibiting STAT3 and NF-kB, resveratrol enhances cancer treatment efficacy and cellular resilience against stress and inflammation. These multifaceted actions position resveratrol as a vital ally in combating aging and improving overall health-span.
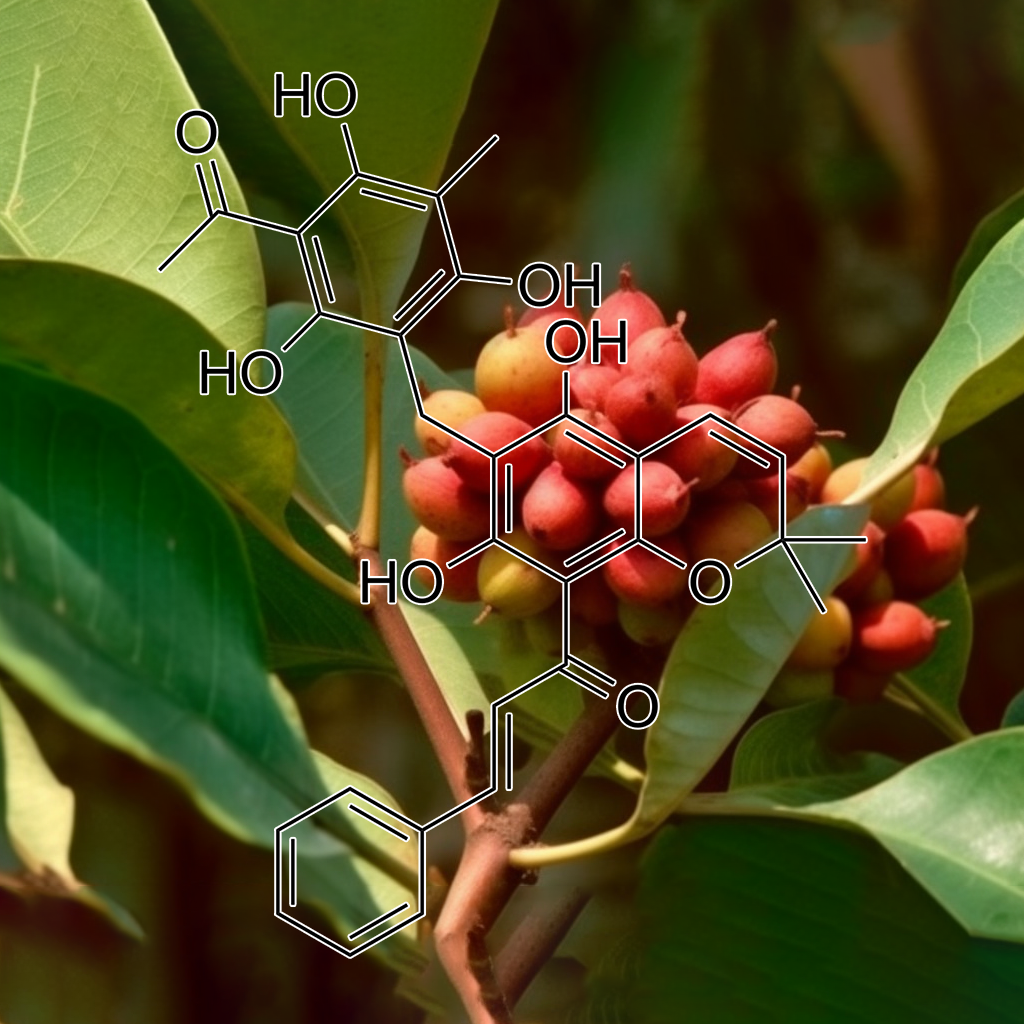
Rottlerin demonstrates potential as a senolytic agent, effectively targeting senescent cells by modulating key pathways such as PI3K/AKT, BCL-2, and mTOR. It induces autophagy and apoptosis in breast cancer stem cells (CSCs) by downregulating anti-apoptotic proteins like BCL-2 and XIAP. Additionally, Rottlerin sensitizes glioma cells to TRAIL-induced apoptosis, enhancing caspase cascades while sparing normal astrocytes. These actions collectively promote cellular health, reduce inflammation, and may contribute to increased longevity.

Rutin, a bioflavonoid, exhibits notable senolytic properties by inducing apoptosis in senescent cells through pathways like PI3K/AKT and mTOR. It effectively reduces the senescence-associated secretory phenotype (SASP), mitigates oxidative stress, and enhances autophagy. In animal models, rutin demonstrates potential anti-aging effects, improving brain function and reducing diabetic atherosclerosis by stabilizing plaques and lowering inflammation. Targeting these biological mechanisms may significantly promote health and longevity by addressing cellular aging processes.

Salvia miltiorrhizae
Salvia miltiorrhizae shows promise in enhancing health and longevity by targeting senescent cells. Its active compound, Tanshinone IIA, induces apoptosis and cell cycle arrest in cancer cells while modulating key senolytic pathways such as PI3K/AKT, BCL-2, and mTOR. Additionally, it inhibits TLR4 expression, reducing inflammation, and supports autophagy and cellular stress responses. These mechanisms provide potential therapeutic benefits in age-related diseases, promoting cellular health and longevity through effective senescent cell clearance.

Scutellarin

Silibinin, a flavonoid from milk thistle, shows promise as a senolytic compound by reducing senescent cells and their associated SASP factors. It modulates key pathways, including PI3K/AKT, apoptosis regulators (BAX, BAK), and Nrf2, enhancing healthspan through autophagy and oxidative stress mitigation. Additionally, silibinin induces apoptosis in prostate cancer by inhibiting Stat3 and suppresses bladder tumor growth by targeting survivin, promoting apoptosis while exhibiting low toxicity in vivo. These findings highlight silibinin’s therapeutic potential for aging and cancer management.
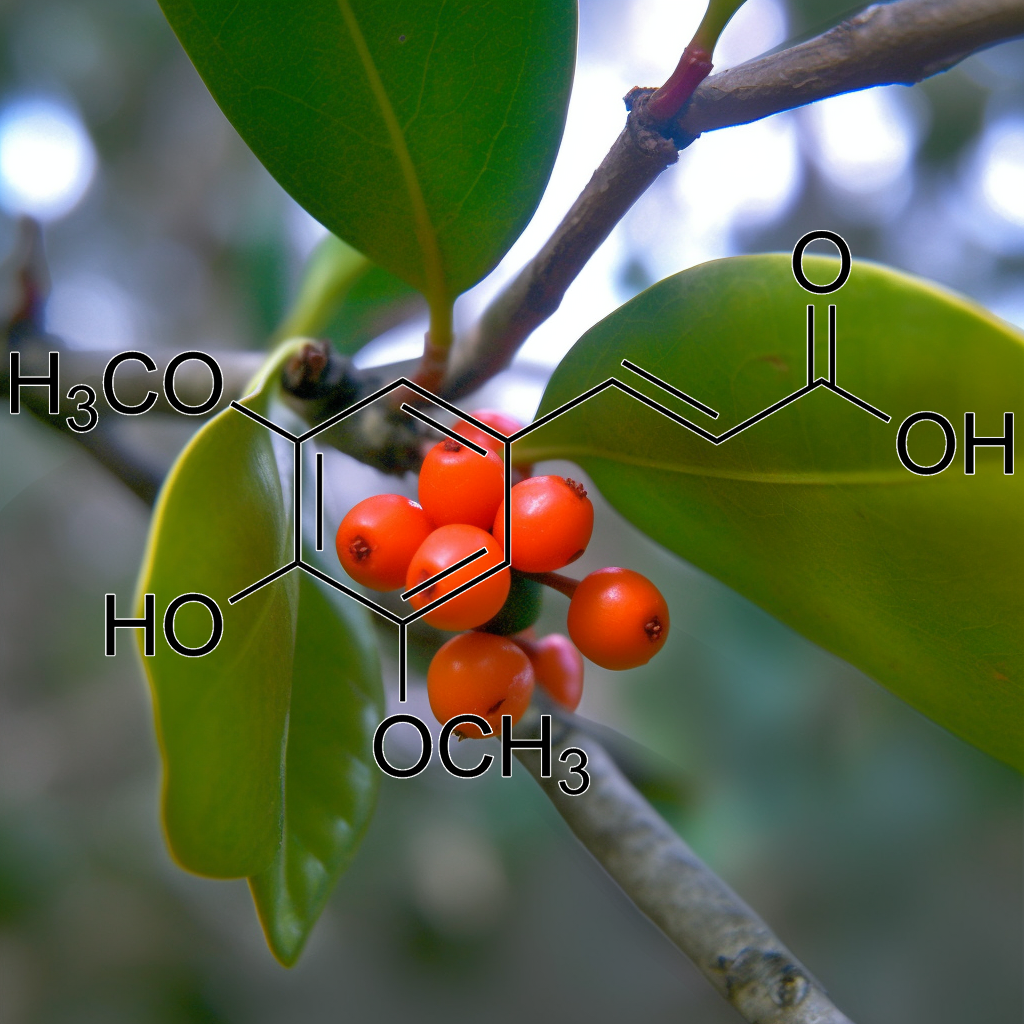
sinapinic acid (Hydnophytum formicarum Jack)
Sinapinic acid from Hydnophytum formicarum Jack exhibits potential senolytic properties by targeting senescent cells, modulating pathways like PI3K/AKT, mTOR, and Nrf2. It influences apoptosis regulators (Bcl-2, Bax), enhances autophagy, and reduces inflammation (NFKB, STAT3), contributing to longevity, healthspan, and combating age-related diseases through cellular rejuvenation. H. formicarum Jack. rhizome extracts, rich in sinapinic acid, exhibit HDAC inhibitory and anticancer activities, inducing apoptosis in HeLa cells, supporting further research for cancer therapy.


St. Rophanthus Gratus (Ouabain)
Strophanthus gratus seed (Ouabain) has potential links to senolytic pathways targeting senescent cells, apoptosis (BCL-2, Bax), inflammation (NF-kB, STAT3), and autophagy (mTOR). It may influence health by modulating cellular stress responses, supporting longevity through pathways like cGAS-STING, PI3K/AKT, and TLR4, enhancing anti-aging interventions. Ouabain demonstrates senolytic and antiviral potential, reducing inflammation, targeting senescent cells, and improving lung function in COVID-19, making it a promising therapy.
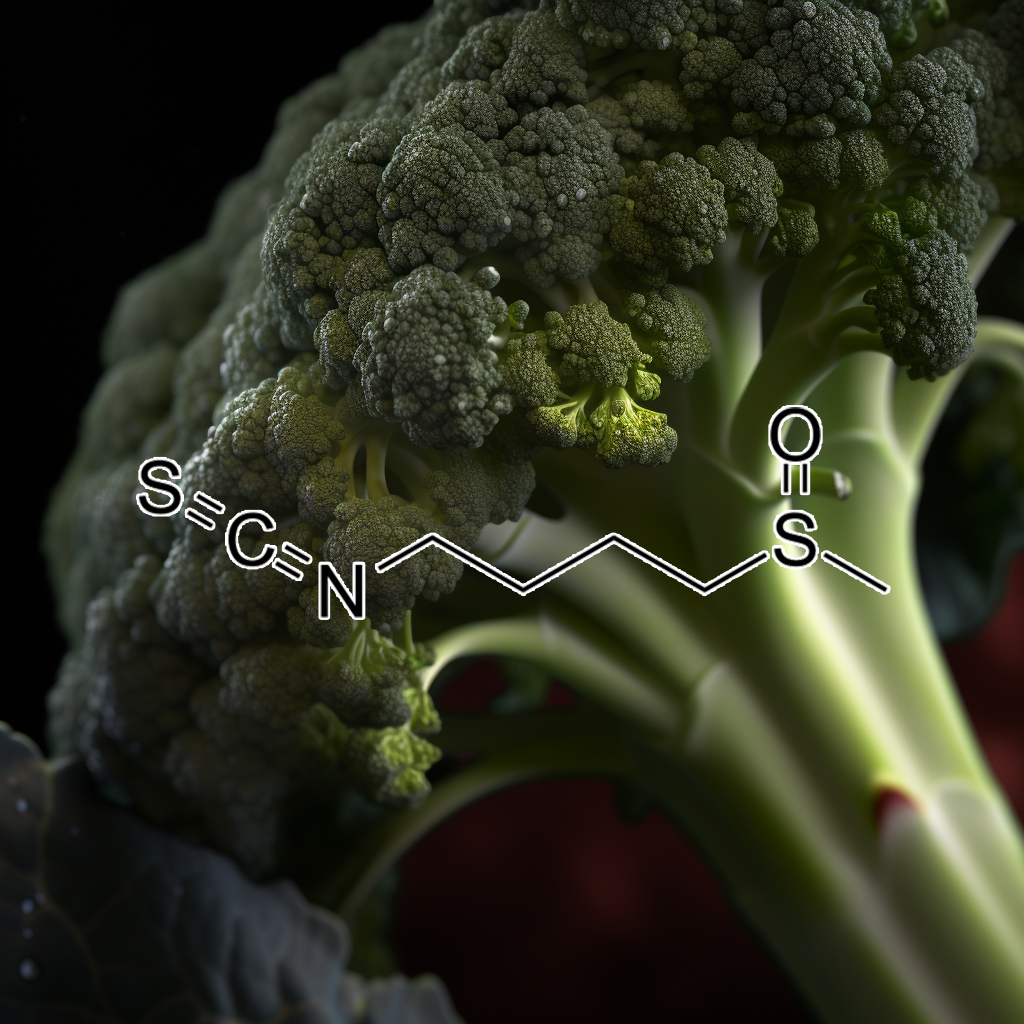
Sulforaphane, a potent compound found in cruciferous vegetables, effectively targets senescent cells, promoting healthy aging and longevity. By modulating key pathways such as Nrf2, mTOR, and apoptosis regulators like Bcl-2 and Bax, sulforaphane enhances autophagy and reduces the senescence-associated secretory phenotype (SASP). Additionally, its role as a histone deacetylase (HDAC) inhibitor supports cancer therapy by improving histone acetylation. Sulforaphane also exhibits anti-inflammatory properties by inhibiting TLR4 signaling, highlighting its therapeutic potential in age-related diseases.

Tangeretin, a potent citrus flavonoid, exhibits senolytic activity by targeting senescent cells and modulating critical pathways such as PI3K/AKT, Nrf2, and mTOR. It induces apoptosis by inhibiting BCL-2/BCL-XL, enhancing Bax expression, and upregulating caspase-3. Additionally, tangeretin reduces senescence-associated secretory phenotype (SASP) markers like TLR4 and NF-κB. By promoting autophagy and cellular resilience, tangeretin supports longevity and health, making it a promising compound in anti-aging research and cancer therapy.

Taraxasterol is a pentacyclic triterpene with promising senolytic effects, targeting senescent cells and modulating pathways like PI3K/AKT, mTOR, and Nrf2. It enhances cellular health by influencing apoptosis through BCL-2 family proteins, including Bax and Bcl-2, and plays a role in autophagy and the senescence-associated secretory phenotype (SASP). Additionally, Taraxasterol shows potential in inhibiting liver cancer cell growth, making it a candidate for cancer therapy and overall longevity improvement.

Thymoquinone, a compound from Nigella sativa, targets key senolytic pathways including PI3K/AKT, mTOR, and cGAS-STING. It modulates apoptosis regulators like Bcl-2 and BAX, reduces SASP, and enhances autophagy. Thymoquinone may improve longevity by targeting senescent cells and influencing pathways such as Nrf2, STAT3, and NF-kB for cellular health and anti-aging effects. Thymoquinone inhibits STAT3 activation, down-regulates survival genes, induces apoptosis, and enhances effects of cancer therapies, showing potential for multiple myeloma treatment.

Ursolic acid, a natural triterpene, exhibits senolytic properties by targeting senescent cells and modulating key pathways, including PI3K/AKT and mTOR. It enhances apoptosis through regulation of proteins like Bax and Bcl-xl while reducing SASP factors such as TLR4 and NFKB. Additionally, ursolic acid inhibits USP7, decreasing cancer cell proliferation. By promoting healthy aging, it helps decrease inflammation and mitigate age-related diseases, supporting longevity and overall health.
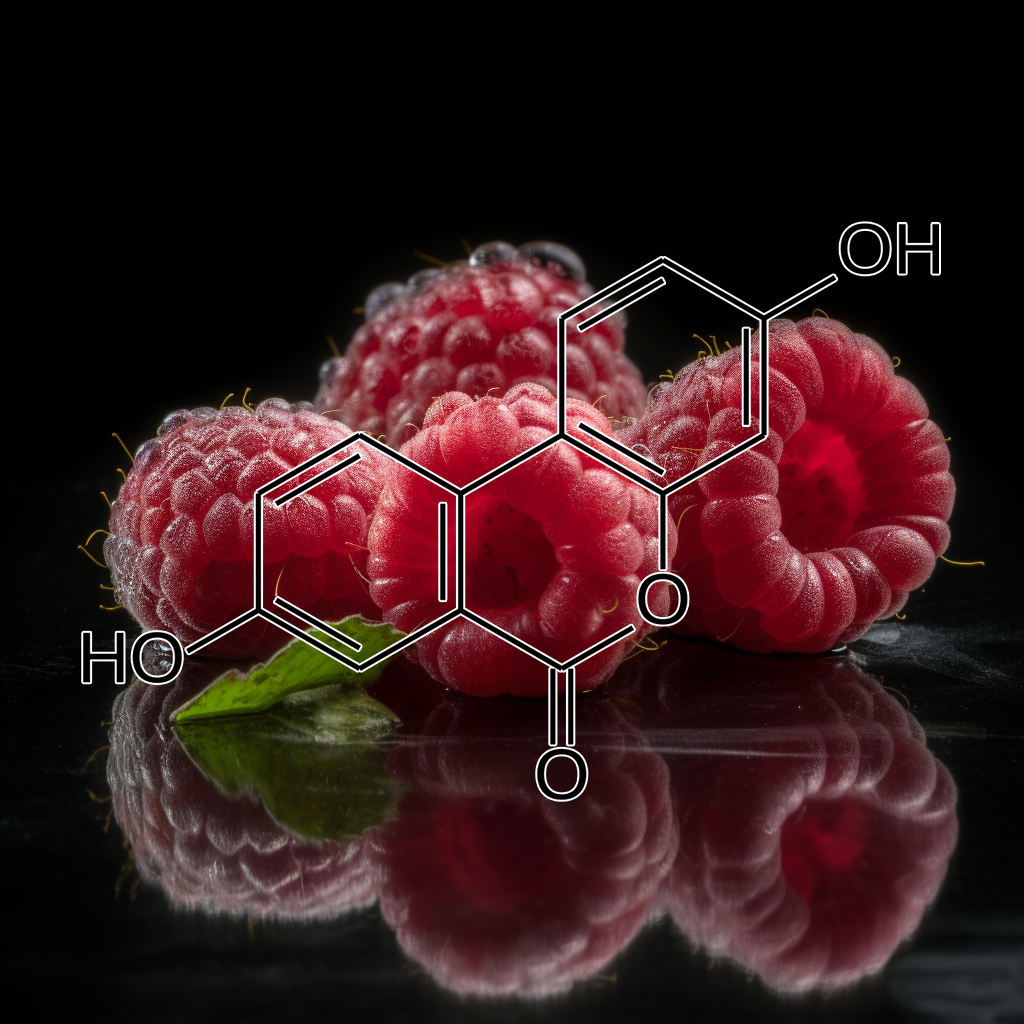
Urolithins are gut microbiota metabolites of ellagitannins; ellagi- tannin-rich foods include walnuts, red raspberries, strawberries, and pomegranates. Since urolithin A (UA) and B are able to attenuate LPS- induced inflammation, inhibiting activation of the NF-κB and Akt sig- naling pathways (Xu et al., 2018), they may emerge as new anti-SASP metabolites. It has recently been noted that oral UA can induce mitophagy both in vitro and in vivo. Urolithin A, derived from ellagic acid, extends lifespan in C. elegans and improves muscle function in mice by inducing mitophagy, dependent on AMPK and mitochondria.
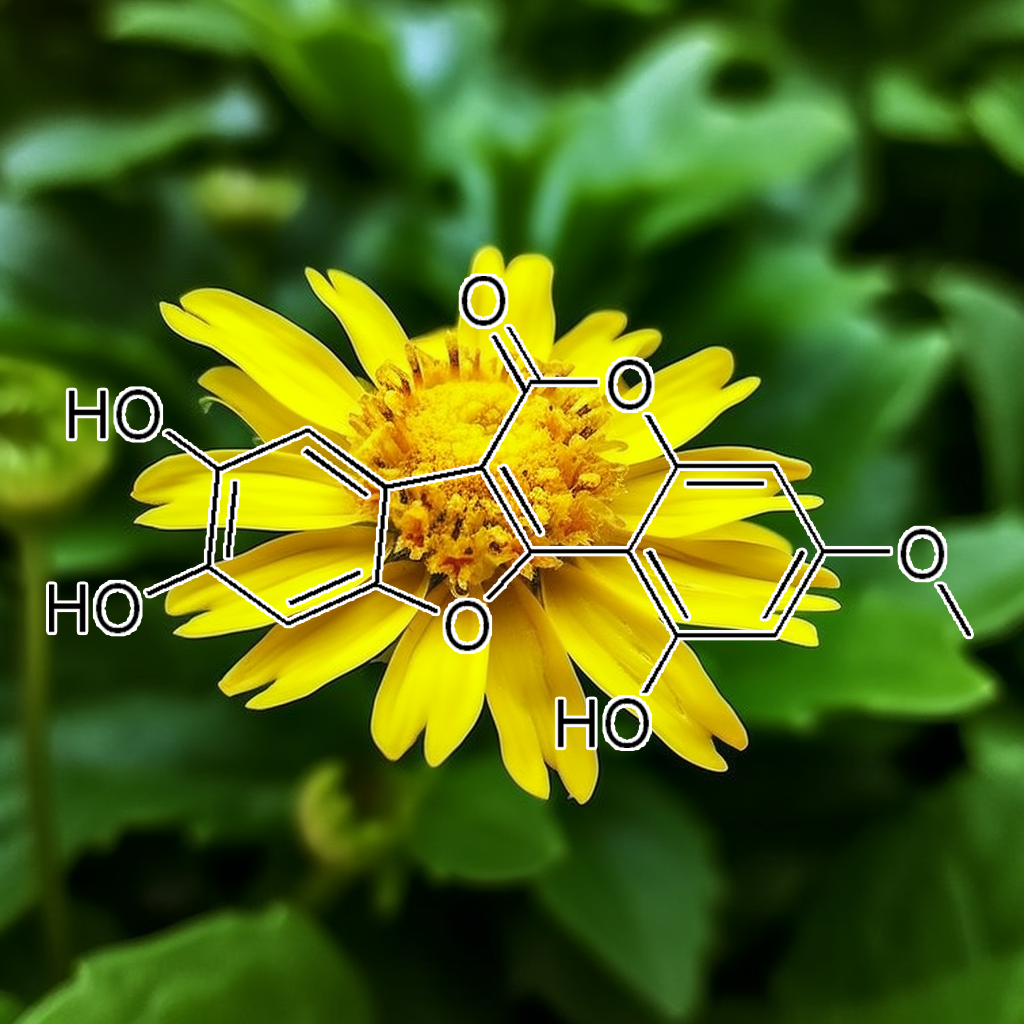
Wedelolactone, a natural compound, is linked to senolytic activity, targeting senescent cells and related pathways like PI3K/AKT, BCL-2, cGAS-STING, and mTOR. It influences apoptosis regulators (Bcl-xl, Bax, PUMA), autophagy, and inflammation (NFKB, STAT3), promoting longevity and reducing SASP, enhancing cellular health, and combating age-related diseases. Wedelolactone (WDL) inhibits melanoma cell proliferation, migration, and invasion by modulating Bax, Bcl-2, cyclin D, p21, and AMPK/AKT signaling pathways in vitro and in vivo.
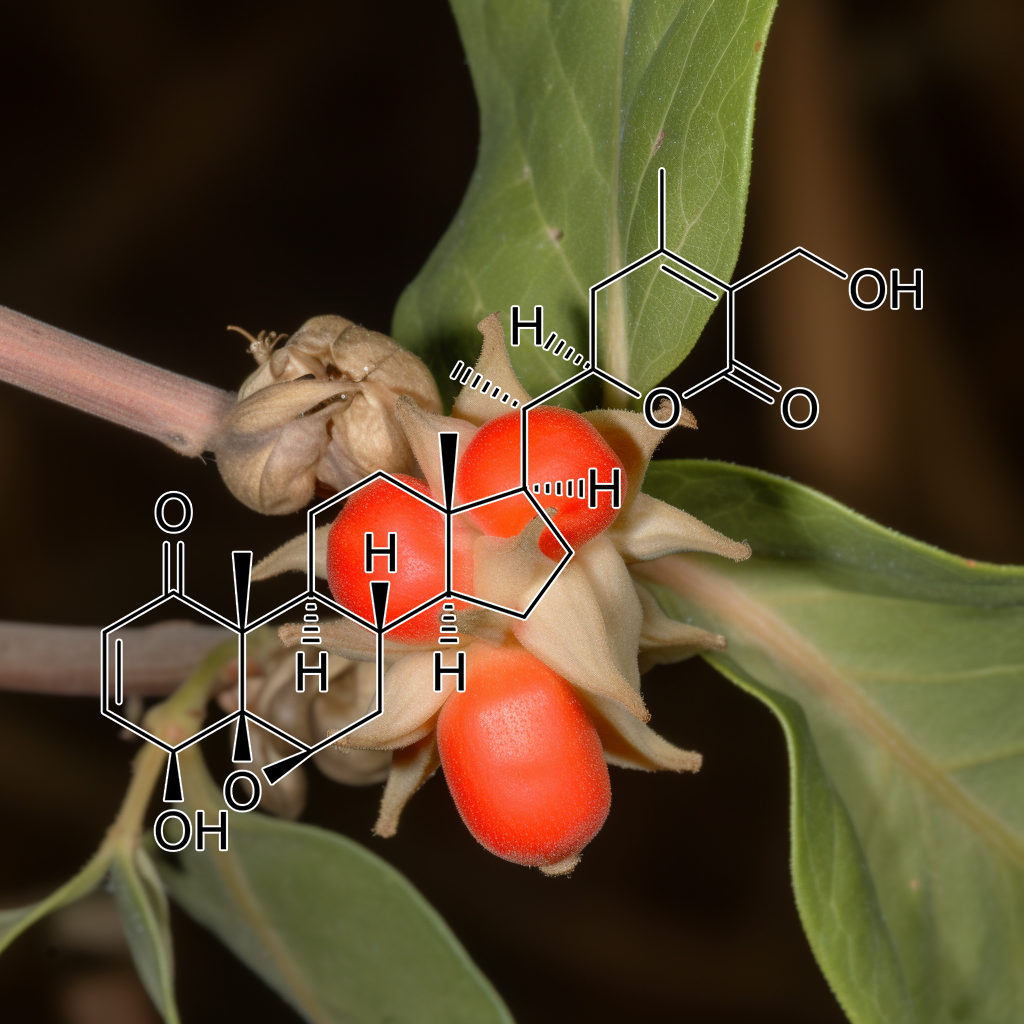
Withaferin A from Ashwagandha exhibits senolytic properties by targeting senescent cells and modulating key pathways such as PI3K/AKT, mTOR, and Nrf2. It inhibits the Senescence-Associated Secretory Phenotype (SASP), enhances autophagy, and activates cGAS-STING, promoting cellular health and longevity. Additionally, its interaction with Hsp90 may improve cancer therapy by disrupting protein maturation. These mechanisms collectively support healthy aging and provide therapeutic potential against age-related inflammation and cancer.

Wogonin is a flavonoid with senolytic properties that selectively targets senescent cells, promoting longevity by modulating key pathways like PI3K/AKT, BCL-2, and Nrf2. It enhances apoptosis and autophagy while reducing age-related inflammation through the suppression of inflammatory markers such as NF-κB and STAT3. Additionally, wogonin inhibits tumor growth and angiogenesis by disrupting VEGFR2 signaling. Its ability to activate caspase 3 and decrease MCL-1 levels highlights its therapeutic potential in cancer and cellular senescence.
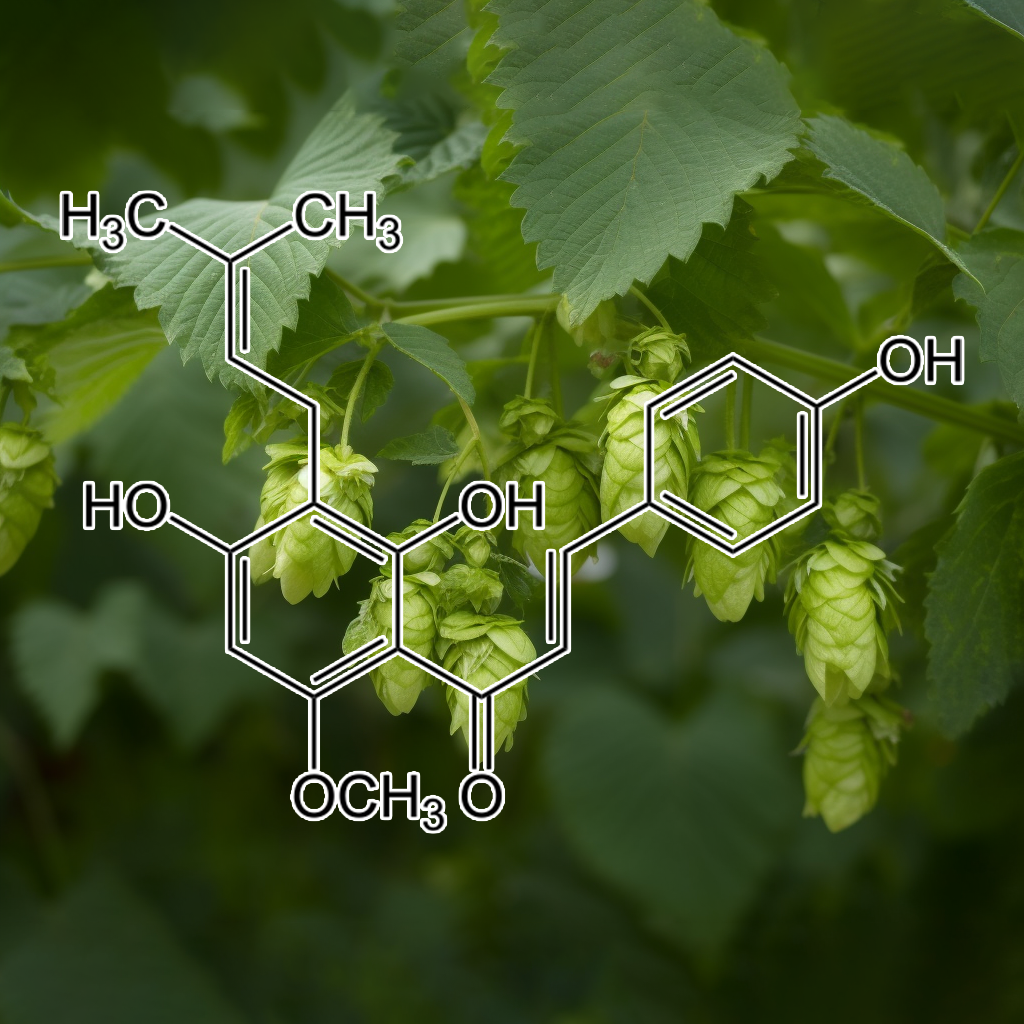
Xanthohumol

Zerumbone, derived from Zingiber zerumbet, exhibits senolytic properties by targeting senescent cells and modulating key pathways such as PI3K/AKT, mTOR, and NF-κB. It enhances apoptosis through the Bax/Bcl-2 ratio and promotes autophagy, influencing SASP factors like STAT3 and TLR4. This compound shows promise in improving health and longevity by reducing inflammation and rejuvenating cellular functions, positioning it as a potential therapeutic agent in age-related diseases and cancer treatments.
| Size | 25g, 50g, 100g, 200g, 300g |
|---|
12 reviews for SENOLYTIC (Zombie Cell Killer)
Related products
-
APIGENIN
$275.00 -
THUNDER (Male Sexual Health)
$275.00 -
LUTEOLIN
$275.00 -
SEVEN SAGES 50:1
Price range: $250.00 through $350.00 -
INTERSTELLAR SHILAJIT
$99.00






Rich Ryan –
As we age more and more of our cells stop working right, and become Senescent or “Zombie” Cells. They just take up space without doing their jobs, and most of them release toxins into the bloodstream as well. Senolytic blend is meant to enable the body into killing these cells off. Knowing this, I was very excited to try Senolytic blend and boy was I not disappointed!
The first thing I noticed was incredible mental clarity. Then after a week, I had to look twice in the mirror, as I started to look younger. I felt less hungry, and more vibrant and alive. Then, and I kid you not, I started winning money at the casino! I’ve been playing around with my intuition and roulette for years, and when I started to take big doses of Senolytic (1/2 tsp twice a day), my intuition sharpened and I started to hit more numbers! To date, I’ve made triple my money back on Senolytic. Not a bad deal eh? Time to order more!! 🙂 🙂 🙂
In my opinion, this is the best blend Interstellar has come out with yet. Really really impressive!!
Teresa Kuhn –
WOW! WOW! WOW! These new blends are the bomb! Gavin, I own every blend you sell except the brand new Anti-Adipogenic. In the last few weeks I added Gynostemma, Helicobacter and Senolytic, and it’s made a huge difference. I started taking your blends last summer, developed major FOMO so bought every one you offer, and I have experienced more energy, less appetite, thicker hair, better focus but nothing compares to the energy and focus that I’ve got today. Fasting for 24 hours is a breeze. Thank you Gavin, you are brilliant, and I am so grateful for all you do and your committment to helping others achieve amazing health and well being.
Alex Tice –
I Am Riding Senolytic Right Now…
⭐️⭐️⭐️⭐️⭐️
As Above, So Below,
The first step to make sure you are preventing a zombie apocalypse on the outside of you is to halt and clear the zombie apocalypse on the inside of you. With this blend, you can kiss the cellular self-sabotage goodbye. In experiencing this Senolytic Blend, I have never felt more like myself ever. We all know this life is a journey where we will continue to travel up our own personally self-constructed ladder to heaven some call Destiny. What they don’t tell us is that the ladder is built inside of us on a cellular, DNA level of spiritually conscious cosmic tapestry that we are. Gavin and the herbs of Nature are helping us go right at the Heart of Healing, tapping into our Innate Unlimited Potential. The Sacred Self-Generating Sanctum is beyond our current imaginative capacity and straight into the Remembrance of what it means to be Human: True Diamond Sun Angelic Unified Broadband Spiritual Stands of Life Eternal.
Symphonic Stardust Expansion.
Gavin is a master of his craft. From purity and quality of selection, to exceptional formulation and design with kyrstalized precision, packaging and professional delivery and service; in every part of the process Gavin and the Interstellar Team leave their mark and open inspiration deep within all participants choosing to go on the inward voyage of a lifetime. As one applies and experiments with the framework that Interstellar has pre-trailblazed for us all, connecting and developing a living relationship with the herbs and the healing forces of Nature comes easier and goes deeper… way deeper !!!
This Senolytic blend will take you through the complete circle of evolution and then up-step you to the next level of your Core Identity and mission purpose of mind, body, and spiritual unfoldment. Whatever you came here to do. Whatever you cant rest from until you become your own living example of Indelible Human Potential Actualized in Full-Empowerment, know that the blends are here as a manifestation of your Eternal Power, and whenever you engage with them, you are in essence taking your power back and returning order, balance, and harmony within the universe. Everything emanates from within, you are your own Source of Power and Truth, you are the Epi-Center and this Senolytic Blend will irrevocably light your fire to StarShine in Authenticity of Your True Eternal Nature.
Stephen Shuman (verified owner) –
Wow!!! This blend is a game changer. I have been using Trinity for months now, and I was worried that maybe I had developed a tolerance to it. The first month on Trinity was amazing, but than I hit a plateau with the results. I would do a fast, but then as soon as I came back onto eating, even if it was on an intermittent fasting keto diet, it was a real struggle to lose the extra weight, especially my spare tire.
I started taking the Senolytic blend a little over two weeks ago with my morning Trintiy in coffee, and have continued intermittent keto and suddenly not only am I losing weight again, but I have a TON more energy for everything in my life!. Not only that, but most of the weight loss is from my gut and love handles. I am down 14.4 lbs so far. Best of all, I am noticing changes in my my body that I was not expecting. My focus is razor sharp. Sex with my girlfriend has not been this good in months. I have been diagnosed with psoriasis, and my skin has not looked this good in years!!
-Stephen Shuman
Mychael Maldonado (verified owner) –
I started takin Gavins blends awhile back and was recommended the sample pack
Which had a variety of different blends I could try out. I tried all of them and my favorites were Peel and Spice and Trinity.
Also Matcha and Pine pollen.
Alot of these blends have very good benefits to your body in a kind of miraculous way.
Its like your body saying WHAT THE HECK WAS THAT! !
In a way like Please give me more.
Every herb and every flower and every bark that is in these herbs treat and feed every cell in your body that is damaged and rebuilds them so that you FEEL BETTER, LOOK BETTER, THINK BETTER, LOVE BETTER..
THEY are amazing and worth your attention.
Peel and Spice have me looking
10 years younger….
Trinity has me and my mind at Peace.
This NEW BLEND SENOLYTIC
Is a BOX Office Smash Hit
It clears your FOG and gives you Mental Clarity, Energy, Focus,
It FEEDS your SOUL
IT IS THE REAL DEAL so to say.
And I combine it with the rest.
It even gives me more endurance and strength…
Its the needed Cherry on top of the whole Cake….thank you
Gavin for your knowledge and help that we all need.
But never make a jump towards
Because we are BLINDED in are way of thinking..and limited…
But with these blends there are no Limitations …
There are no I CAN’TS
ONLY I WILL AND I CAN.
Take that Leap Now……
You won’t have any regrets….
Sebastian Gaeta –
This was the first interstellar blend I have tried. I didn’t really know what to expect with this. Im in my early thirties and generally very healthy and take a lot already, so I didn’t know if I would notice anything. I wasn’t sure how many senolytic cells I had or what the difference would feel like. That said I barely took any of the ingredients listed here before so I was intrigued. I wasn’t fully fasting but trying to be as keto as possible.
I started to really look forward to taking it, I found there was a new level of clarity I was finding myself in most days without realising. Also I found synchronicities increasing drastically and strange situations and items manifesting, sometimes quite literally large items that I had been looking for endlessly with no success turning up on my doorstep. Looking back some very big plans have been set in motion in this period and important projects finished. Things are just coming together quite effortlessly where as some were dragging on for months with no resolution before.
Very interested to see if this continues from where it left off when I try this again or if this effect diminishes now I’ve finished my supply!
Either way definitely a new convert to Interstellar! It looked the part from the ingredients list and certainly is.
Rochelle Vitler (verified owner) –
I started using Interstellar Blends about nine months ago and since then I can honestly say I have not had a single day where I’ve felt sick or had any sort of sniffle, cough or cold. Every day I am mentally alert, calm and physically healthy.
About three months ago I added Senolytic Zombie Cell Killer to the daily mix of Trinity, Super Hair, Autophagy and either Nebula or Thermo.
Senolytic seems to have cleared up a niggle in my left knee and a dodgy lower back that I’d accepted as part of getting older. Neither gives me any trouble anymore. Fasting is also easier than ever.
The most exciting difference for me with Senolytic though has been that my 50-year-old body, which was flabby from losing 35 kg probably a bit too quickly (even though I exercised the whole time) is starting to tone up. I thought that might not happen and was prepared to accept the weight loss and deal with the soft, squishy body but the changes are exciting and very much welcome. I’ll certainly be keeping Senolytic in the mix.
Thanks Gavin for these awesome blends.
Debra Ferguson (verified owner) –
I’ve been dealing with health issues for 7-8 years now. When they started, I was around 35 – lifting weights and jogging every day, feeling like I was aging like a fine wine to…. breaking my health somehow. It most likely due stress at the time. I was crazy Type A personality but have worked diligently on my mind and spirit since then so now I’m just as cool as the Fonz 24-7. But I’ve never really gotten back to full health. I’ve seen 17 doctors since then. My symptoms are in line with auto immune issues and NO ONE CAN TELL ME WTF is WRONG.
I have chronic swelling of the hands/fingers, hairloss, joint pain in my hips, mystery skin issue on my torso that isn’t visible but feels like really tiny, rough patches of skin, muscle tightness and pain sometimes so severe even having a 4 hour deep massage doesn’t bring it relief, mystery rash (peri oral dermatitis) that comes and goes, inflammation in my esophagus, sleep issues and even though I exercise daily, my calves have this weird rippling like cellulite but it’s not? I don’t know. It’s weird.
Ultimately, I think the chronic inflammation is at the root cause of all my issues as I know when it’s so rampant in your body, your cells cannot perform. But after 38 different labs in the last 5 or so years, the only thing that shows up is high eosinophils levels which can be linked to allergies or parasites (been tested for both – negative – and even treated for parasites regardless). The levels are not SO high that it sets off the alarm bells though. Everything I mentioned above sometimes leaves me depressed and ready to call it a life. The chronic pain is exhausting. I don’t take meds. Just deal with it.
I’ve been to a gastroenterologist, dermatologist, rheumatologist, naturopath, functional medicine practitioner, DUMB ASS FUCKING REGULAR WESTERN MEDICINE DOCTORS… I’ve done every diet know to man, including a 30 day water fast. Ketosis is definitely something that allows me to feel “better” but still not 100%. I will not give up until I get my health back. If something doesn’t work, I will try something else. Most recently, I did a genetics test and it showed that I’m predisposed to have a hard time creating the elements necessary to detox the body. My body struggles to create glutathione and its precursors as well as with autophagy. I always feel more energetic when I fast and that really explains why.
As you can imagine with all this stuff going on, I’ve had to empower myself with knowledge of all kinds of things because useless doctors can’t help. I used to follow Cole and the Snake Diet but wanted to learn more about dry fasting particularly and that’s when I stumbled upon Gavin’s site. I was, ummm, blown away by the information and after going down the rabbit hole for 2.5 hours, felt like I had leveled up riding a rainbow unicorn towards some new solutions for what’s ailing me. Gavin makes Cole look like romper room. I immediately unfollowed the Snake Diet and signed up for the Interstellar FB group.
Anyhoo – I started with Spice and Peel based on Gavin’s recommendation to treat inflammation, some Hypnotic to address the sleep issues and a sample of the Nebula to see if it would affect my overall mood/mindset. I kind of lack confidence and wanted to give no fucks all the way around. The Hypnotic had me sleeping through the night and feeling rested WITHOUT taking any other non-natural sleep aids. The Nebula certainly had me feeling just relaxed and comfortable in my own skin. Very hard to explain. Like I always have this negative storyline running in the back of my mind and that went away. The Peel and Spice didn’t seem to have as strong as an effect on my inflammation as I had hoped but I also didn’t get the 200:1 concentration which will be my next purchase.
After seeing some measurable results and doing more research on Gavin’s site, I decided to try the Helico. I don’t have stomach issues (very grateful for that) but a few of the studies I read seemed like it couldn’t hurt to try it. I also decided to order the Autophagy Activator and Senolytic based on my genetic report data. Here’s what I experienced:
I usually hop on my treadmill from anywhere from 25-45 minutes. Due to the health stuff, I don’t really have a lot of energy. I work from home and it’s easy to be sedentary. I power through what I can. After taking the combo of Gavin’s blends, my walks went from 25-45 minutes to a minimum 60 minutes to 90. It’s like, even if I want to give up, I’m so focused, I can’t. THIS IS NUTS! I love it. Today, I even increased the difficulty and got my heart rate up about 20 BPM from my normal walks and still did over an hour. Unbelievable. I had less inflammation in my hands when exercising (it usually gets really bad). And I don’t know if it’s related but I had a crazy vision of breathing in golden light and exhaling black air. Almost like whatever negative energy or dysfunction/dis-ease I’m experience was being replaced with white light energy. I can’t really explain it, but it definitely happened. The weird skin issue on my torso is mostly healed. No rash on my face.
I’ve only been taking the Autophagy and Senolytic for 2 days as I’m doing a 7 day fast! Can’t wait to see what the rest of the week looks like! I am telling everyone about the blends. I’m sure I sound like a lunatic but that’s ok. This is the longest testimonial I’ve ever written. If you’ve made it this far, congratulations and buy some blends. You won’t be sorry!
Dr. Lynn Peters (verified owner) –
During the Summer of 2020 I did a 134 day fast with no food, only the blends. This was a journey to my own health maximization and couldn’t have been done without the blends. How do I know this? In 2018 I did a 110 day fast before finding Interstellar Products and I am able to make an educated, objective comparison.
I am a trained healthcare Clinicians and completed this fast while in a pandemic, working full time in s hospital and had no illness or sick days. My mind was focused and clear and I experienced no exhaustion or hunger throughout the 4.5 months. This I attribute to the blends.
I prepared for this fast and had been purchasing blend specials for months, so I had them on hand. I started with the Ultimate Sampler. That is a huge bargain and a great way to try what amazing products are here. Yes, they cost more, but hospital bills, doctor visits, medications and daily pain and suffering in a diseased body cost exponentially more. My quality of life matters and fasting saves you money on all of those things AND no grocery bills.
Initially I took every blend, every day. That got me into the habit of fasting and helped my body break the habit of food. I had plenty of fuel in my fat cells, but the body needs to adapt to not being overfed. I listened to my body and reduced the quantity of daily blends to Senolytic, Super Hair, Peel and Nebula as my base each day. I would then add in Autophagy, Trinity, Spice, Shilajit and others. THEY ALL have their place in my life. I still take blends daily and will for my lifetime.
The customer service is bar none, the quality of the products is outstanding and the intangible benefits to a healthy life are beyond my vocabulary.
Fasting is a choice. It is a proven strategy for longevity and maximizing health. Interstellar Blends, supercharge fasting results by supporting your biology with scientifically supported ingredients that boost your systematic ability to reset, reverse and heal all the damage accumulated during your lifetime. Our lifestyles in the “modern” era have wreaked havoc on the anatomy and physiology of the human structure. Interstellar Blends are the antidote to a life filled with dis-ease and damage.
Choose you. Choose a healthy future living your best life with health and vitality. There is not a blend here that will not have a noticeable impact on you for the better.
I am a customer for life. Fasting will forever be a part of my days and blends will always be included so that I am ensuring my body has what it needs to repair for the onslaught of environmental and other toxins that we encounter each and every day. Interstellar Blends are like internal cellular armor protecting your system from attack. You owe it to your mind, body and Spirit to support your vessel with the best. Interstellar Blends are the top of the line and your vessel will thank you.
Nicole Rodriguez (verified owner) –
The interstellar blends are absolutely life-changing. In my 30s and after having kids, my body changed; weight loss was harder, finding time for exercise became difficult to find too. So, I let the weight pile up and found excuses on why it was not the right time to focus on changing my life. Well, as I approached 40, the weight kept increasing, and health issues were added to my already worrisome focus on the climbing scale. However, the health issues really changed my perspective.
I tried so many different products, putting time & money into each; and, nothing worked. Then I found these amazing blends. I started with thermo & then added into autophagy & senolytic. I was amazed, because not only was I losing weight, I felt good inside too. I then started the weight loss combo package to ensure I was getting all the proper blends to support this healthy-living journey – and wow, did those blends work wonders. I have lost 50 lbs on these blends over the course of a few months, and what is most important is I feel good too. I have been losing my hair due to other health issues, so I recently added in the super tonic hair blend too.
Finally, I am so happy to report the best news so far that after returning to the Dr.’s recently, all of my health issues are gone – the numbers reverted back to what they were a few years ago. Thus, I plan to keep up this weight loss journey & healthy living. I have also added stationary bicycling, walking, and light strength workouts to my routine. Again, this was not just about weight loss for me, but more about living longer for my family, being happier for me, and being in a more healthy state for all of us. These blends are life-changing and worth the price you pay.
Akshita –
This Senolytic blend is a game-changer. I have been using it with black coffee twice a day for 15 days and I have already lost 5kgs without doing any strict diet. Although this blend is capable of a lot of transformation and I look forward to using it routinely.
One other change that I noticed was a drastic increase in my energy levels. Before, I always struggled to complete my tasks or get up from my bed. I was always anxious.
But in just 15 days, I am seeing a lot of difference in my energy levels. I am more energetic and less anxious. To be honest, I have not added any exercise regime in my routine. Thank you Gavin for these blends
Kristin Mazza –
OKAY this product takes the cake. Absolutely unbelievable. So I am usually a repeat user of Niagra and Trinity for their benefits. However, my cravings and my weight gain were just out of control. Well not after starting this product. I followed the instructions to the T and saw amazing results after about a week. I wasn’t over eating any more, I had way less cravings for sugar and foods where before I had no self control. I even kicked my late night snack habit just because this zapped the hunger away. The overall result after a few weeks of eating right, the right portions, and exercise I lost about 8 lbs and have since kept the weight off. If anyone is trying to figure out something new to introduce to their routine because you feel nothings working, this might be your new go to! I will definitely be ordering again next time I need a body reset.